

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
Molecular Modeling and Structure-based Drug Design Systems
Click on the individual program names to access to their detailed features.
![]()
Homology Modeling Professional for HyperChem, Revision H1
with Gaussian Interface for HyperChem & ONIOM Interface for Receptor
Compatible to NAMD Molecular Dynamics and ABINIT-MP / GAMESS Fragment Molecular Orbital Quantum Mechanics Calculations for Entire Molecular System
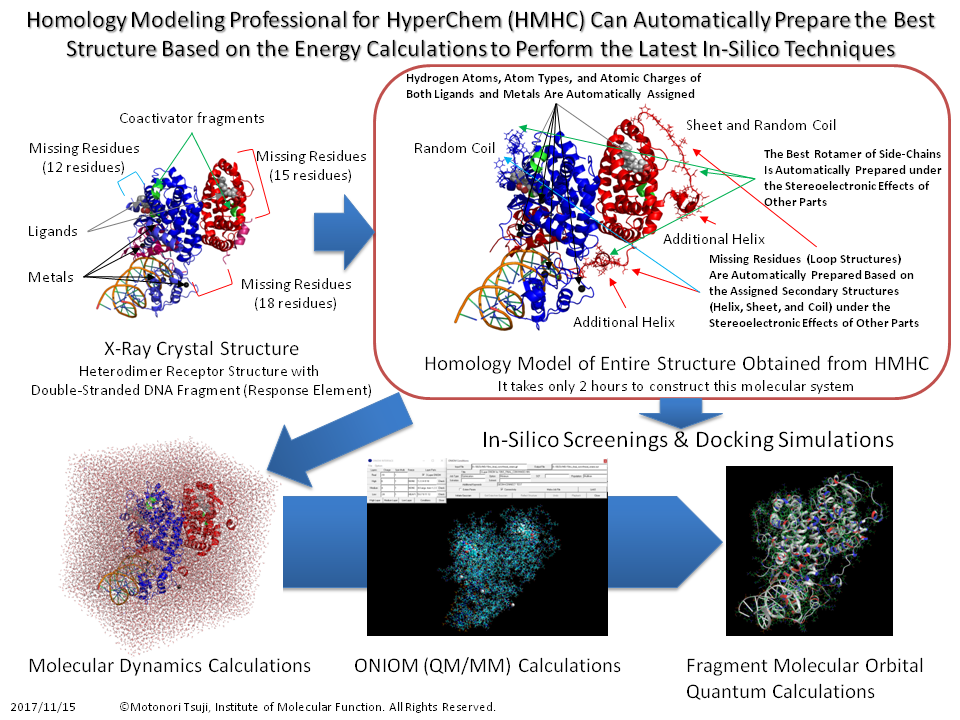
The Latest In Silico Drug Design Platform: Evolution of Homology Modeling Professional for HyperChem, Revision H1.
Strengthened quantum mechanics calculation program, Gaussian,ONIOM Interface, compatible to file format of molecular dynamics calculation program, NAMD (VMD: CHARMM-based PDB), compatible to file format of fragment molecular orbital calculation programs, ABINIT-MP (BioStation Viewer) and GAMESS (Fu /Facio)
In quantum mechanics calculations for the entire structure of big molecular system such as biomacromolecular system, the single point calculations for the initial structure obtained from classical molecular mechanics calculations and/or classical molecular dynamics calculations cannot converge or will give abnormal energies. For obtaining reliable results from quantum mechanics calculations such as fragment molecular orbital methods of entire molecular system, the initial structure must be prepared using geometrical optimization calculations via ONIOM methods. ONIOM Interface for Receptor can provide the best solution for the quantum mechanics calculations of the entire molecular system of which the precise initial structure is prepared from Docking Study with HyperChem and Homology Modeling Professional for HyperChem.
Motonori Tsuji, et. al. FEBS Open Bio., 7, 391-396, 2017, DOI: 10.1002/2211-5463.12188.
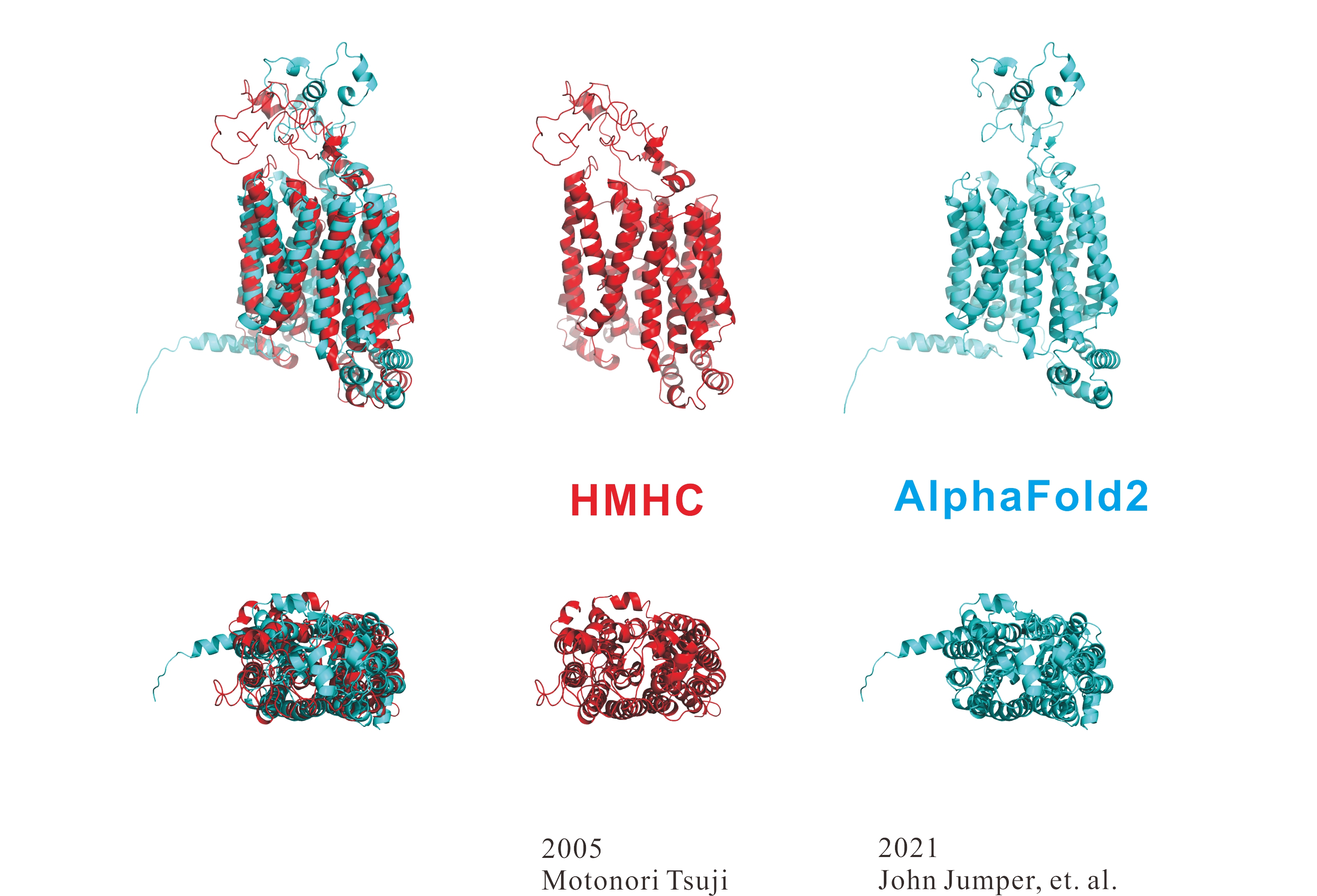
AlphaFold2 vs Homology Modeling Professional for HyperChem (HMHC)
The following figure shows the results of three-dimensional structure prediction for an unknown protein sequence for 20 years including family proteins, using HMHC and AlphaFold2.
HMHC can provide a precise three-dimensional structure since HMHC can model the side-chain rotamer conformations and the interactions between other bioamacromolecules and small molecules as well as crystal waters.
Homology Modeling Professional for HyperChem is the latest molecular modeling package.
The latest method on the basis of the energy calculations via HyperChem and Gaussian programs.
It can carry out the molecular modeling, functional analysis, and simulations of a big molecular system using comprehensively the ab-initio quantum chemistry calculations and the molecular dynamics simulations as well as the molecular mechanics calculations.
The program package can be used for performing the protein homology modeling in the presence or absence of the small molecules, water molecules, other biological molecules, metal atoms, and the small molecule covalently bound to the model under the vacuum or the various solvated conditions. The package supports the state-of-art technologies for modelings, simulations, and analyses.
The protein homology modeling can be performed using only three module programs to optimize the structure of the main chain, side chain, and whole molecule system. Since these module programs can automatically operate, the homology modeling can be precisely performed under the almost full automatic conditions.
Powered by Homology Modeling Professional for HyperChem THE IMPOSSIBLE BECOMES POSSIBLE.
New, High-Performance Software for Protein Modeling, Functional Analysis, and Simulation.
Homology Modeling Professional for HyperChem is the latest molecular modeling package which can carry out the molecular modeling, functional analysis, and simulations of a big molecular system using comprehensively the ab-initio quantum chemistry calculations and the molecular dynamics simulations as well as the molecular mechanics calculations. The program can control seamlessly both HyperChem and Gaussian packages and can be simultaneously fed back their calculation results to the big molecular system in the HyperChem graphical user interface. The whole molecular system can be operated in a quantum chemistry via the full-automatic ONIOM interface to Gaussian. All operations are fully automated in the GUI-based manner, and thus the program supports the logical molecular modeling, functional analysis, and simulations in the life science research such as the structure-based drug design, as well as an unexplored research. Moreover, this program package can cooperate with a protein- and ligand-flexible docking and screening program package bearing the next generation drug discovery technology, PIEFII technology. From the Revision B1, the protein modeling, functional analysis, and simulations can be performed in the level of figure of a literature or a demonstration using the full-automatic rendering and labeling functions.
Pamphlet of Homology Modeling Professional for HyperChem, Revision H1 (PDF: 1 MB; Japanese)
Pamphlet of Homology Modeling Professional for HyperChem, Revision G1 (PDF: 4 MB; Japanese)
Pamphlet (Japanese; PDF: 12 MB) for earlier version
SBDD Pamphlet (PDF: 2 MB; Japanese)
References
Motonori Tsuji, Molecular Science. 1, NP004, 2007.
Science. 319, 624-7, 2008.
Biochemical and Biophysical Research Communications. 339, 173-177, 2010.
Biochemistry. 49, 10647-10655, 2010.
Plant Cell Physiol. 53, 1638-1647, 2012.
Motonori Tsuji, Journal of Structural Biology. 185, 355-365, 2014.
Motonori Tsuji, et. al., Journal of Computer-Aided Molecular Design. 29, 975-988, 2015
Motonori Tsuji, Journal of Molecular Graphics and Modelling.62, 262-275, 2015.
Motonori Tsuji, et. al., FEBS Open Bio. 7, 391-396, 2017. DOI: 10.1002/2211-5463.12188.
Motonori Tsuji. Journal of Computer-Aided Molecular Design.31, 577-585, 2017. DOI: 10.1007/s10822-017-0025-6.
J. Pharmacol. Exp. Ther. 372, 277-284, 2020.
Motonori Tsuji, FEBS Open Bio 10, 995-1004, 2020.
J. Virol. 94, e00252-20, 2020.
Chem. Pharm. Bull. 68, 1193-1200, 2020.
PLOS ONE 16, e0257705, 2021.
Motonori Tsuji. Int. J. Mol. Sci. 23, 11009, 2022.
The most powerful and sophisticated molecular modeling environment
HyperChem front-ended
The most powerful computational chemistry environment
Gaussian back-ended
Homology Modeling Professional for HyperChem Outline
Homology Modeling Professional for HyperChem is a package that consists of some module programs which are required for performing the protein modeling, functional analysis, and simulations using HyperChem which is well-known molecular modeling software in the world. The individual module programs in the package carry out new technologies. Therefore, by the fusion of these technologies to the HyperChem functions, the package serves as the latest structure-based drug design system in the drug discovery as well as serves as the latest protein modeling, functional analysis, and simulation systems.
Software Development for Researchers
Homology Modeling Professional for HyperChem is designed to reflect intuitive operation. That is, almost all of purposes can be performed only clicking the provided button in the individual module programs. Moreover, it is only necessary to operate a rotation of molecules in the HyperChem workspace, since several tools conferred from the package can automatically navigate the operations. For example, the program distinguishes between proteins, waters, and other molecules, and shows differences of these molecules. All module programs support the full-automatic rendering and labeling functions and thus the protein modeling, functional analysis, and simulations can be performed in the level of figure of a literature or a demonstration. The "Undo" function is available when the structure is modified. An objective part is automatically centralized in the HyperChem workspace. Thus, it is possible to make homology modeling of proteins without any knowledge for the HyperChem operations, although power users can carry out all functions of HyperChem.
Applicability for the Complicated Molecule System
Homology Modeling Professional for HyperChem covers almost all of the PDB structural data registered to date, since the package can deal with about 100,000 atoms (HyperChem 7.5 or later). The protein modeling as well as the protein functional analysis can be carried out under all conditions, where the molecule system can contain small molecules, metals, and polymers, etc. Moreover, the protein modeling can be performed using our new technology, even though the above molecules and atoms covalently bind to protein molecules. Their atom types are assigned automatically, and their atomic charges can be generated using many theoretical calculations. Otherwise, it can also give manually. Thus, the package can support the high-level, structure-based drug design such as the lead optimizations, since the package can analyze the detailed interactions between compounds such as seed and lead compounds and protein molecules such as the highly-precise protein models and the crystal structure optimized by this package.
The Most Powerful Computational Chemistry Environment
Homology Modeling Professional for HyperChem provides a powerful protein functional analysis environment, since this package supports all theoretical calculations integrated into HyperChem. The package can also use those supported in combinations of Mopac2000 and Mopac Interface for HyperChem and those supported by Q-Chem for Windows, if these programs are available. Moreover, the package can automatically reflect the electronic state of all molecules except for protein molecules to the corresponding molecules in the HyperChem workspace via Gaussian Interface for HyperChem. The whole molecular system in the HyperChem workspace can be operated in a quantum chemistry via ONIOM Interface for Receptor.
Other Features
No preparation and editing are necessary for parameter files and PDB files. Of course, a detailed log file including the used parameters and the results are generated automatically. The individual module programs in the package carry out a file manager function. The early versions of HyperChem can be renewed as the latest protein modeling and functional analysis environment, since the package runs on HyperChem 5.x, 6.x, 7.x, and 8.0x.
Powerful Performance
Moreover, Homology Modeling Professional for HyperChem shows the powerful performance which is able to support logically an unexplored study such as the ligand recognition mechanism of receptor. Since this package runs on the Intel based Windows platforms, large advantage of both computational cost (computational speed) and user interface is expected in comparison with those of the corresponding system in the Unix platforms.
Reproducibility and Logical Molecular Modeling
Homology Modeling Professional for HyperChem can provide the reproducibility and logicalness of the models by performing molecular modeling on HyperChem consistently. As the results, the package enables large shortening of total operations, as well.

See Homology Modeling Tutorial using Homology Modeling Professional for HyperChem.
See also Technical Supports for the latest Test Information and Release Notes.
Module Programs
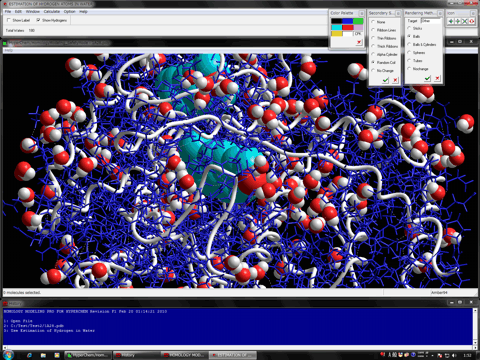
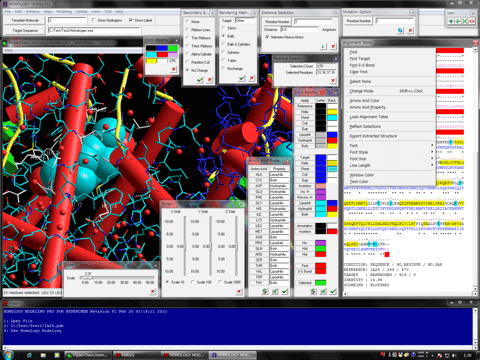
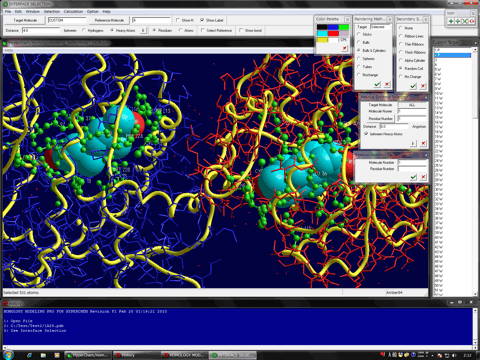
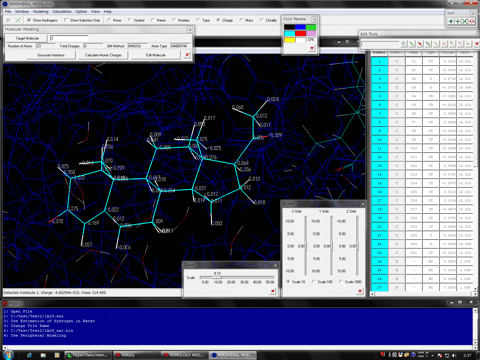
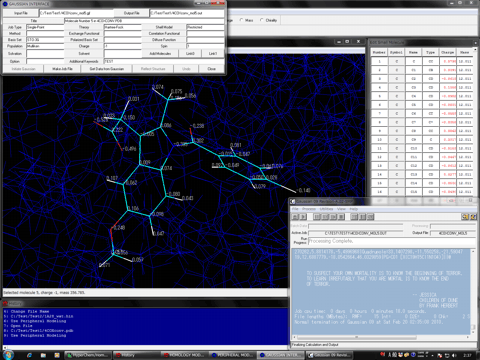

Estimation of Hydrogen Atoms in Water
Homology Modeling Professional
Peripheral Modeling Professional
Side Chain Rotamer Modeling Professional
Gaussian Interface for HyperChem
Recommended Minimum System Requirements
Processor: Multi-Core Intel Core or Xeon 2 GHz (Intel Pentium III or later)
Operating System: Microsoft Windows 10 and 11 (Microsoft Windows 95 or later, XP is necessary for all function)
Memory (RAM): 8 GB (Minimum 256 MB, ONIOM calculations need 64 GB or more)
Monitor: SXGA or more
Video Card: OpenGL board recommended
Mouse: Multi-functional mouse
Other: HD 512 GB or more; CD-ROM drive; Network card
Software Requirements
Essential
HyperChem (Windows version; except for Student version): HyperChem Release 8.0.10 Professional for Windows (Release 5.1.1 or later, Release 7.5.2 is necessary for all function)
TclPro1.2 (Windows version): TclPro1.2 for Windows is necessary to run Homology Modeling Professional for HyperChem. TclPro1.2 is available on the Web free of charge.
Option
Gaussian: Gaussian16 64bit multi-processor version (recommended) (Gaussian03, 09, or 16 (single- or multi-processor version) is necessary to run Gaussian Interface for HyperChem and ONIOM Interface for Receptor).
OpenBabel (64bit or 32bit Windows version): OpenBabel for Windows is necessary for exporting in MDL SDF, Tripos MOL2, or AutoDock PDBQT format from HIN (HyperChem format) or PDB format.
Other
NAMD and VMD is necessary for performing ns scale molecular dynamics. ABINIT-MP and BioStation Viewer or GAMESS and Fu orFacio can be used for performing quantum mechanics calculations for entire molecular system.
Description
Compatible to the Protein Data Bank format versions 3.0-3.3.
Homology Modeling Professional for HyperChem is a package of Tcl/Tk programs (mainly user interface) and 32bit Windows console applications.
These programs are compiled by the TclPro1.2 compiler. Thus, in order to run the package, you must install TclPro1.2 in your Windows system, prior to the installation of the package.
A substantial Online-Help (English) is available from individual programs in the package.
The package includes 3 user's manuals (presently Japanese Language Only).
Manual Contents (Japanese Language)
Manual 2 Contents (Japanese Language)
Reference Manual Contents (Japanese Language)
Important: When you installing the software, please make sure that HyperChem and TclPro1.2 have been installed in your system.
This package does not include HyperChem and Gaussian.
* HyperChem is a registered trademark of Hypercube, Inc.
** Gaussian is a registered trademark of Gaussian, Inc.