![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
有機合成化学者のための論理的ドラッグデザイン
PIEFII技術で可能になる論理的ドラッグデザイン手法 - Drug Design Methodology
−ドラッグデザインワークベンチとしてのHomology Modeling for HyperChemおよびDocking Study with HyperChem−
2006年9月26日
有機合成化学者のための論理的ドラッグデザイン Download PDF
ミミックデザイン
生物活性物質をデザイン、合成するにおいて、メディシナルケミストリーやアグリカルチャラルケミストリーの合成研究室でいまなお日常的に行われている手法は、天然リガンドあるいはリード化合物の平面構造を鉛筆でなぞり、その構造の上で一般的な合成フラグメント(シントン)同士をつなぎ合わせ、合成が単純で経験的に毒性の低そうな化合物でミミックしようとするものである。
図1に、このミミック手法でレチノイドXレセプターに対する合成リガンドをデザインする一例を示す。
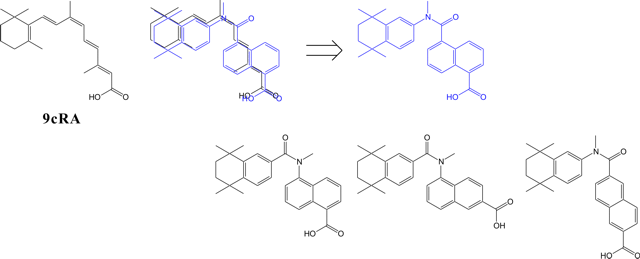
図1.9-cis レチノイン酸(9cRA)の平面構造を利用したミミックデザイン
この手法では最初にデザインした化合物(ミミックスまたはアナログ、図1の上段)を基に、さらに類似構造(図1の下段)をもつ化合物を複数デザインして合成、アッセイするのが定石である。
こうしたデザインが日常的に行われる理由はその手軽さと、これまでは、このようなデザインでも合成例がなく、つまり新規性があり、また、遠からず活性が示される場合があるからである。
図2に最初にデザインした化合物の合成ルートを示す。
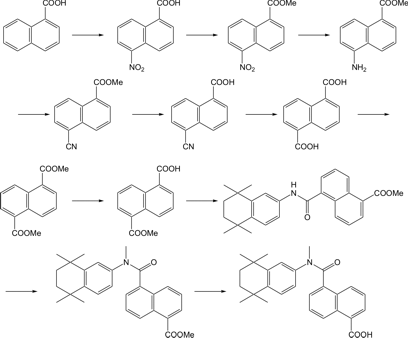
図2.9cRAアナログの合成ルート
単純な構造ではあるが、実際の合成には合成の熟練者でも数週間から1か月を要する。また、試薬代や精製に必要な溶媒代、アッセイ費用、人件費等を考慮すると、1化合物あたりに必要な時間と経費は膨大である。
さらに、10年前であれば、こうしてデザインした化合物は確かに新規と考えられるが、現在では、安易にデザインした化合物はすでに既知であるばかりでなく、同じ目的で合成されている確率が極めて高い。既知でない場合には、むしろ目的活性がないためだろうと疑うべきである。
なお、こうしてミミックデザインした4種類全ての化合物にRXR活性はなかった。
ドラッグデザイン序盤に弱い構造ベース創薬
ミミックデザインは短時間でデザインでき、天然リガンドやシードまたはリード化合物の平面構造と若干のレトロ合成(逆合成)に関する知識以外を必要としない代わりに、先に述べたように、今後いろんな意味でリスクの高い手法になりつつあるといえる。
逆に、バイオインフォマティクスの分野ではポストゲノム時代を迎え、膨大な数のタンパク立体構造情報が蓄積されつつある。また、ホモロジーモデリングなどの手法により立体構造の不明なタンパクについても既知立体構造(鋳型)を利用して論理的に、簡単にモデリングできるようになってきている[1]。
このような中で、必然的に構造ベースのドラッグデザインが注目されている。
しかしながら、構造ベース創薬に代表されるドッキングスタディーなどのシミュレーション技術はデザインした後の化合物あるいは既存化合物を対象とした主にドラッグデザインの中盤以降の技術と考えられ、ドラッグデザインの序盤で簡便、論理的に化合物をデザインする手法とはいえない。
これに対して、ドラッグデザインの序盤で、より個々の有機化学者に近い立場から、構造ベースで薬物設計する方法論としてデノボデザインがある。しかしながら、デノボデザインの方法論を支えるこれまでの理論や技術が未熟であったため、デノボデザインは全くといってもよいほど利用されてきていない。
なお、リガンドベースのQSAR技術はリード最適化のようなやはりドラッグデザイン中盤以降でのデザイン手法である。また、HTSや同じ概念を持つバーチャルスクリーニング技術などはドラッグデザイン序盤での生物活性物質のデザインを必要としないかわりに、特定の大企業などでしか実際には実施不可能であり、個々の研究者が合成テーマをはじめに持つ大学研究機関などでは相容れない手法といえる。
シームレスな構造ベースドラッグデザインを可能にするPIEFII技術
したがって、ミミックデザインのような簡便さをもちつつ、より論理的な手法を提供できる技術が構造ベースドラッグデザインの序盤に不可欠であるといえる。
これを解決できる技術が、PIEFII技術である[2]。
以下にPIEFII技術を搭載する構造ベース創薬支援システムを用いた実際のドラッグデザインについて述べる。
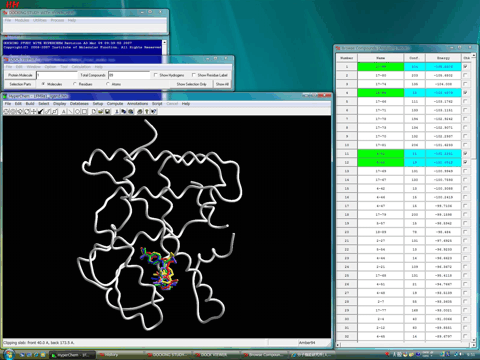
図3はX線結晶構造解析されたアゴニストコンフォメーションを採用するRXRαレセプターリガンド結合領域の立体構造である。
図3.hRXRαリガンド結合領域
これを基に、RXRαレセプターに対するアゴニストをデザインする。また、本レセプターの立体構造が不明な場合は、ファミリータンパクの立体構造を鋳型にホモロジーモデリングを実施して得たモデルを用いても良い。
タンパク立体構造に対してPIEFIIプログラムを実行すると、瞬時に外部因子の重原子が占有可能な位置およびその位置における相補的な物理化学情報が点(相互作用点:ファーマコフォア)として表示される[3](PIEFII技術については詳細ページを参照されたい)。
図4は予測された相互作用点(ファーマコフォア)と実際に複合体を形成している9cRAの構造(赤のスティック)を同時に示したものである。また、メッシュはリガンド結合部位のファンデルワールス表面を示している。なお、予測された相互作用点(ファーマコフォア)情報(緑色は疎水的な重原子、黄色は静電的(負電荷)な重原子が占有可能であることを示す)はリガンド結合部位のみならず、天然リガンドの骨格構造、化学的性質を忠実に再現している。
図4.PIEFIIアルゴリズムによりhRXRαリガンド結合部位に予測される構造ベースファーマコフォア情報
研究者は予測された各相互作用点(ファーマコフォア)の物理化学的性質を満たしつつ、かつ各点を重原子で占有するように図5のように合成リガンドをデザインするだけである。また、提供されるリガンド結合部位形状や周辺残基情報を加味することで天然リガンドやリード化合物の骨格にとらわれない、新規な骨格をイメージしやすくなる。
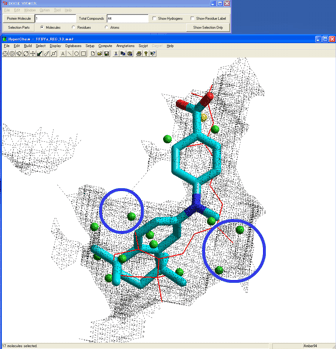
図5.予測ファーマコフォア情報を利用したリガンド結合部位上での合成リガンドの設計
ちなみに、図6のようなミミックデザインを行う有機合成化学者はいないであろう。
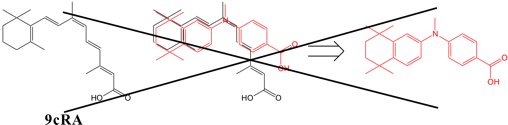
図6.ミミックデザインの限界
以上の作業だけでも有用なデザインが可能であるが、さらに実際にデザインした化合物が理想的に複合体を形成できるかどうかをドッキングシミュレーションで予測することができる。
まず、天然リガンド(9cRA)がこの立体構造中に含まれている9cRAの複合体構造を再現できるドッキング条件(再ドッキング)をタンパク・リガンドフレキシブルドッキングを用いて決定する[3]。なお、この場合は天然リガンドとの複合体の立体構造が既知であると仮定しているが、天然リガンドとの複合体の立体構造がない場合にも、リーズナブルなドッキングモードを与える条件かどうかを判定することは様々に可能である。
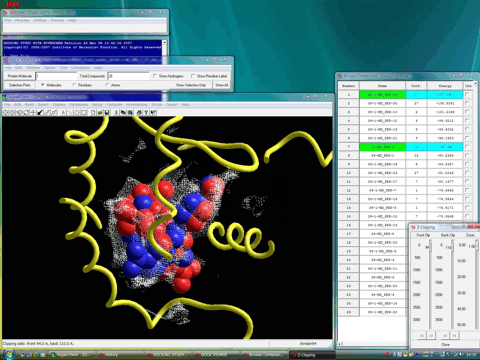
本ドッキング条件にて複数の複合体構造が得られるが、図7に示した複合体構造(緑色のチューブ)はその中で最も安定な相互作用エネルギーを採用したものである(図中の表は相互作用エネルギーの大きい(安定)ものから順にソートされている)。図7では、実際の複合体構造と同時に示している。
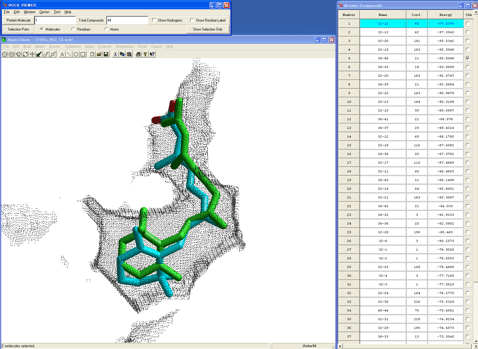
図7.生体高分子−およびリガンドフレキシブルドッキングによる再ドッキング
次に、研究者はPIEFIIを用いてデザインした化合物の平面構造さえ用意すればよく、後はプログラムによってこれを自動的に三次元化し、同条件にてドッキングシミュレーションのスタートボタンを押すだけでよい。
図8はこうしてデザインした化合物のドッキングシミュレーションの結果である。非常にリーズナブルな複合体構造(赤色のチューブ)が予測され、この最安定複合体の相互作用エネルギーについても天然リガンドの相互作用エネルギーに匹敵している。
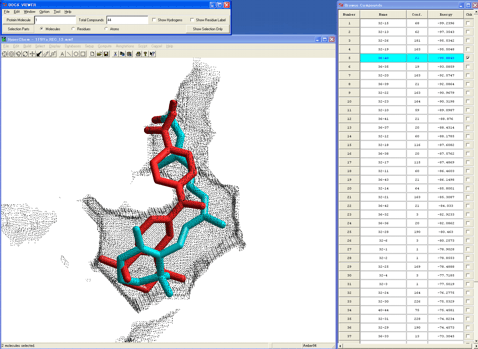
図8.デザインした化合物の最安定複合体構造
表1に実際のアッセイ結果について示す。赤色の行が本化合物についての活性である。
表1.ジフェニルアミン誘導体(DAシリーズ)のRXR活性[4]
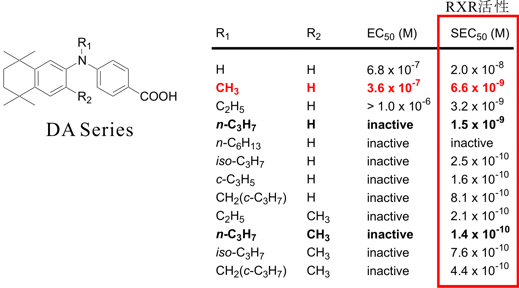
今回のデザインでは図5の青色の円で示した部分の相互作用点(ファーマコフォア)情報は利用しなかったが、上のアッセイ結果からも明らかなように、これら相互作用点(ファーマコフォア)を占有するであろう化合物、すなわち、R1がメチル基のかわりにn-プロピル基、R2にメチル基を導入した化合物、に最高の活性が認められている。このことはまた、本手法がドラッグデザイン中盤以降のリード最適化などにおける構造修飾のための指針としても利用できることを示唆している。
安定複合体を形成した化合物に最高被占軌道(HOMO)や最低空軌道(LUMO)などの分子軌道(図9)、電荷密度、あるいは静電ポテンシャル(図10)などの電子状態を同時に様々な形式で表示し、研究者のインスピレーションを最高に掻き立てることができる。
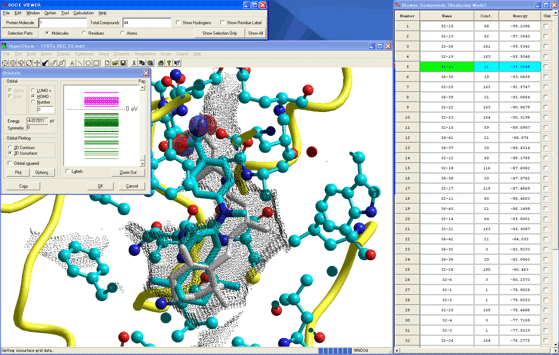
図9.最安定複合体構造を採用するジフェニルアミン誘導体の最高被占軌道
HOMOやLUMOは化合物の毒性と相関があるし、相互作用解析にも役立つ。また、安定複合体のコンフォメーション下にある化合物自身の安定性が予測できる。こうした多彩でかつデモンストレーションレベルの描画を完全自動で作成できることも本創薬支援システムの特徴であり、他の既存システムとは一線を画す。
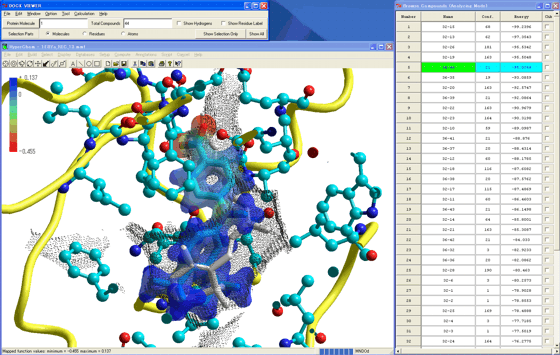
図10.最安定複合体構造を採用するジフェニルアミン誘導体の静電ポテンシャル
なお、同条件にて、最初にミミックデザインした化合物についてドッキングシミュレーションを同様に実施したところ、本化合物との複合体構造(青色のチューブ)は図11の1複合体構造のみしか得られなかった。
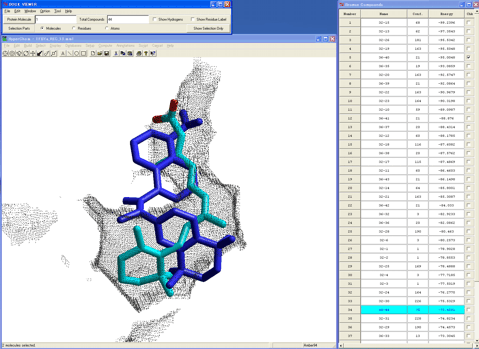
図11.ミミックデザインした化合物のドッキングシミュレーション結果
結果、ミミックデザインした化合物は天然リガンドである9cRAとは全く異なった複合体構造しか採用できず、相互作用エネルギーも20kcal/mol以上低い(弱い)ことが予測され、リガンド結合部位形状とを考慮して、本化合物はRXRに結合しないと容易に推測できる(図12)。
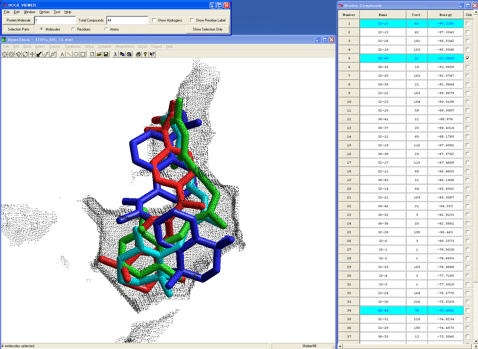
図12.構造ベース予測ファーマコフォア情報を用いてデザインしたジフェニルアミン誘導体(赤)とミミックデザインした化合物(青)および複合体構造中の9cRA(シアン)の同時表示
以上のシミュレーションは準備作業を含めても半日程度で終了できるため、ミミックデザインのハイリスク、合成とアッセイにかかる時間と経費を考慮すれば、デザインの段階で、本手法を実施しておくことの意義は極めて大きいといえる。
さらに、本手法を用いることで、ポストゲノム創薬、特に構造ベース創薬を志向したドラッグデザイン、分子設計がその序盤から、より論理的に、そしてストーリー性をもって実施できるようになる。
[1]. Motonori Tsuji. Homology Modeling Professional for HyperChem, Revision A3, 2006, Institute of Molecular Function, Saitama, Japan.
[2]. Motonori Tsuji. Seitaikoubunnsi niokeru sougosayoubui no yosokuhouhou.2006, Patent 2006-125188.
[3]. Motonori Tsuji. Docking Study with HyperChem, Revision A2, 2006, Institute of Molecular Function, Saitama, Japan.
[4]. Kiminori Ohta, Motonori Tsuji, et al. Potent Retinoid Synergists with a Diphenylamine Skeleton. Biol. Pharm. Bull., 1998, 21, 544-546.
2006年9月26日
製品紹介論文
Motonori Tsuji. Molecular Science, 1, NP004, 2007.
成果学会報告
成果論文
Microbiol. Spectrum. 12, 2024.
J. Autoimmun. 139, 103085, 2023.
Motonori Tsuji*. Int. J. Mol. Sci. 23, 11009, 2020.
PLOS ONE 16, e0257705, 2021.
Chem. Pharm. Bull. 68, 1193-1200, 2020.
J. Virol. 2020.
Motonori Tsuji*. FEBS Open Bio., 10, 995-1004, 2020.
J. Pharmacol. Exp. Ther. 372, 277-284, 2020.
Motonori Tsuji*, J. Comput. Aided Mol. Des., 31, 577-585, 2017, DOI: 10.1007/s10822-017-0025-6.
Motonori Tsuji*, Koichi Shudo, Hiroyuki Kagechika. FEBS Open Bio., 7, 391-396, 2017, DOI: 10.1002/2211-5463.12188.
Motonori Tsuji*, J. Mol. Graph. Model., 62, 262-275, 2015.
Motonori Tsuji*, Koichi Shudo, Hiroyuki Kagechika. J. Comput. Aided Mol. Des., 29, 975-988, 2015.
Motonori Tsuji*, J. Struct. Biol, 185, 355-365, 2014.
Chem-Bio Informatics Journal, 12, 1-13, 2012.
Plant Cell Physiol. 53, 1638-1647, 2012
Biochemistry. 49, 10647-10655, 2010.
Biochemical and Biophysical Research Communications. 395, 173-177, 2010.
Science. 319, 624-627, 2008.
Homology Modeling Professional for HyperChemおよびDocking Study with HyperChemはPCベースで個人用創薬ワークベンチとしても利用できる非常に優れた次世代の構造ベース創薬支援システムです。
本システムを用いたその他の論理的リガンド設計のページが利用可能です。
受託研究・共同研究も受け付けています。お気軽にお問い合わせください。
|
MFDDインシリコ創薬受託研究サービス 詳しくはこちら |