![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
分子モデリング、ドラッグデザイン、インシリコ創薬支援システム:タンパク質−リガンドドッキング・核酸−リガンドドッキング・タンパク質−ペプチドドッキングソフトウエア
![]()
真のフレキシブルドッキングシミュレーション&高速バーチャルスクリーニング/インシリコスクリーニングプログラムパッケージ
Docking Study with HyperChem, Revision H1
Essential, Premium Essential, Professional, Advanced, Ultimate, and Cluster
with AutoDock Vina In Silico Screenings Interface
Compatible to rDock, SMINA, and GNINA Docking Simulations, NAMD Molecular Dynamics, and ABINIT-MP / GAMESS Fragment Molecular Orbital Quantum Mechanics Calculations for Entire Molecular System
- 世界初、革新ドラッグデザイン技術 PIEFII搭載 Drug Design System -
![]()
![]()
![]()
![]()
AutoDock, AutoDock Vina, rDock, RxDock, SMINA, GNINAドッキングスクリーニング互換機能搭載
DSHC:新型コロナウイルスに対する世界初大規模仮想スクリーニング成果を達成、国内最初の抗SARS-CoV-2薬医薬品候補リスト論文発表を達成(2020年5月1日)
DSHCを用いた受託研究サービスがご利用いただけます。多数の実績がありますので、是非導入前にご検討ください。
最新情報 最新バージョンは8.1.5(2026/7/4) 追加機能と今後のロードマップを公開
DSHCが搭載する複数のドッキングアルゴリズムに加え、AutoDock、AutoDock Vina、rDock, SMINA, GNINAのドッキングアルゴリズムも組み合わせて精密な分子ドッキングからバーチャルスクリーニングまでサポートする最強のドッキングプログラム
rDockなどSDF形式でドッキング結果を出力するドッキングプログラムとの互換性機能を追加。rDockやSMINA・GNINAドッキング結果の解析、閲覧、絞り込み機能を搭載。rDockで一次スクリーニングを実施し、得られたドッキングモードを初期構造としてAutoDockまたはAutoDock Vinaでの二次スクリーニングが実施でき、さらにSMINAやGNINAと連携。
Docking Study with HyperChem, Revision H1がインシリコ創薬統合プラットフォームとして更に進化。
AutoDock Vinaバーチャルスクリーニング完全対応(化合物データベース作成−ファイル準備−実行−閲覧−絞り込み全自動、Docking Studyモジュールプログラムの結果と同時閲覧も可能)
分子動力学計算NAMD(VMD)CHARMM-basedファイル互換機能追加、フラグメント分子軌道計算ABINIT-MP(BioStation Viewer)/GAMESS(FU/FACIO)ファイル互換機能追加
最先端のインシリコ創薬において、生体高分子システムなどの巨大分子システムに対して全系量子力学計算を実施する場合、分子力学計算による構造最適化計算や分子動力学計算で用意した初期構造では一点計算さえ収束しないか、異常なエネルギー値を与えます。フラグメント分子軌道法などで全系量子力学計算を実施する場合は、ONIOM法で予め初期構造を更に構造最適化して置く必要があります。ONIOM Interface for ReceptorはHomology Modeling Professional for HyperChemやDocking Study with HyperChemで準備した高精度初期構造(複合体構造)を量子力学計算用に更に精密化して全系量子力学計算を成功させる最善のソリューションを提供します。
Motonori Tsuji*, Koichi Shudo, Hiroyuki Kagechika. FEBS Open Bio., 7, 391-396, 2017, DOI: 10.1002/2211-5463.12188.
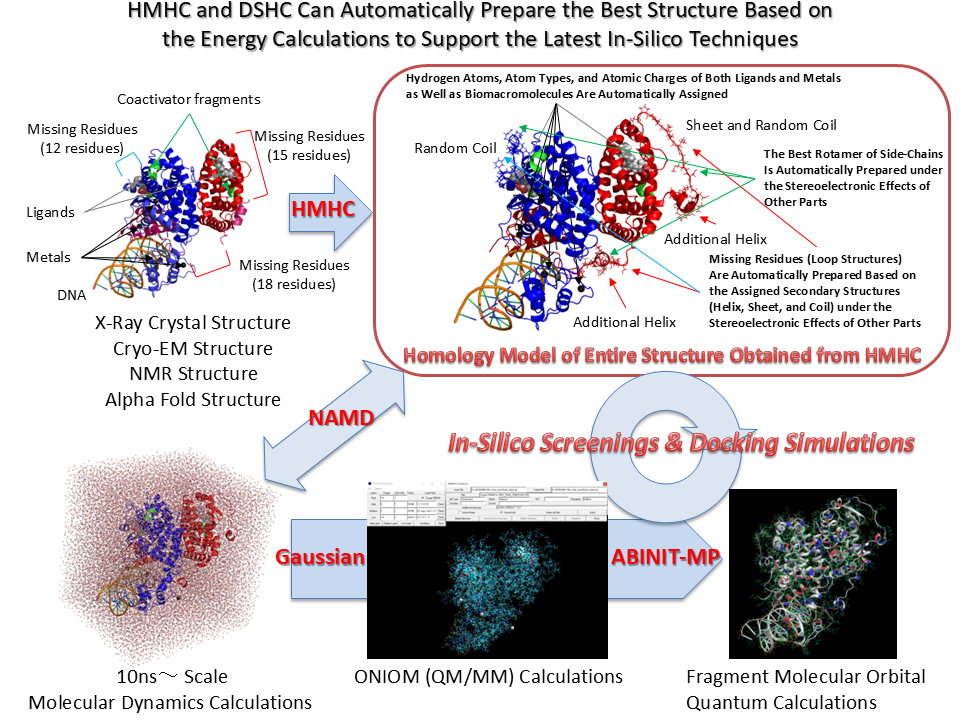
HMHCのONIOM法でリガンド結合部位の電荷と構造を調整した場合はDSHCでもONIOM電荷と構造を反映させてドッキングできます。DSHCではコンフォメーション毎に任意半経験分子軌道法で電荷をアサインしながらドッキングできる機能が搭載されており、半経験的ではあるものの量子化学ドッキング、量子化学インシリコスクリーニングがサポートされる世界で唯一のシステムです。
統合分子設計支援システムHyperChemで全自動生体高分子-リガンドフレキシブルドッキングシミュレーション、バーチャルスクリーニング
様々な状態の生体高分子システム(タンパクおよび核酸分子システム)に対応
従来製品にはない非グリッドアルゴリズムを採用し、HyperChem高信頼性高速計算エンジンによる全系を対象とした高精度手法
構造ベース予測ファーマコフォア点情報を利用する独自のドッキングアルゴリズムPIEFIIによる高信頼性を誇る安定複合体構造探索手法
ドッキング・スクリーニングに要求される各種従来機能に加え、生体高分子システムの誘導適合効果を考慮したフレキシブルドッキング機能、アポ体へのドッキングなど誘導適合効果を超えた大きな構造変化にも対応したフレキシブルドッキング機能、標的生体高分子以外の高分子、低分子、水分子、金属原子、共有結合置換基などからの立体電子影響下でのフレキシブルドッキング機能、試行化合物のコンフォメーション毎に任意半経験分子軌道法による電荷アサイン機能、United AtomおよびAll Atom条件の様々な組み合わせ機能、溶媒和条件下ドッキング機能、リスタート機能、分散処理機能など従来製品では未踏の最先端手法を駆使でき、精密なドッキングシミュレーションからin silicoスクリーニングをサポートします。さらに、プロフェッショナルユーザーは分子力学計算・量子力学計算パラメータからドッキング・スクリーニングパラメータに至る全パラメータをGUIベースで詳細に自由に調整できます。
誰でもすぐに使いこなせるほど簡単操作で高精度なドッキングシミュレーションからバーチャルスクリーニングが実施できます。
Docking Studyモジュールプログラムが搭載する非グリッドアルゴリズムによるAll Atom条件及びUnited Atom条件での生体高分子・リガンドフレキシブルドッキング、リガンドフレキシブルドッキング、剛体ドッキング、部分構造ドッキングなどの各種ドッキングアルゴリズムに加え、AutoDock Vinaインシリコスクリーニングインターフェイスを搭載し、AutoDock Vinaによるグリッドベースのドッキング・インシリコスクリーニング機能が同時に利用できます。rDockやSMINA・GNINAのドッキング結果も閲覧・絞り込みでき、これらのドッキングアルゴリズムを様々に組み合わせる手法を提案しており、実際に正解複合体構造が得られることを複数の学術論文で発表しています。
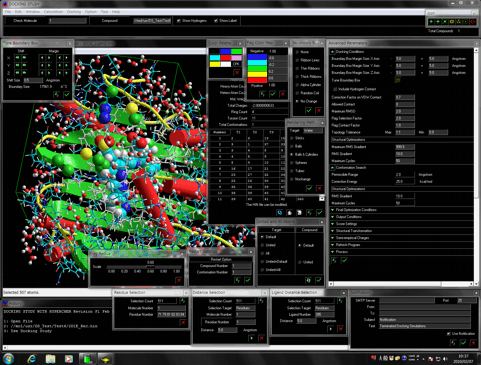
かつてない高性能、高機能、高速、完全GUIベース、全自動生体高分子・リガンドフレキシブルドッキングシミュレーションプログラム
Docking Study with HyperChemでは、Homology Modeling Professional for HyperChemで作成した高精度生体高分子モデルあるいは精密化した既知立体構造と、活性化合物または医薬候補化合物との結合様式を相互作用エネルギーの観点から推定し、リード最適化などの高度な創薬研究を支援することが可能です。
完全自動で主観をなくした網羅的ドッキングが可能で、生体高分子システム(タンパク質分子システム並びに核酸分子システム)およびドッキングに用いる化合物ともにフレキシブルに取り扱える、真のフレキシブルドッキングプログラムです(既存のフレキシブルドッキングプログラムの大部分は化合物のフレキシビリティだけしか考慮できません)。化合物に対しては網羅的なコンフォメーションサーチを実行できることはもちろん、生体高分子の側鎖や主鎖のフレキシビリティ、生体高分子システムに含まれる水分子、低分子、金属原子などのフレキシビリティを考慮したドッキングも極めて容易で、誘導適合効果(インデュースドフィット効果)などもまともに取り扱えます。
パラメータファイル等の編集作業やコマンドライン作業を一切必要としません(ヘビーユーザーは自動作成される詳細なパラメータファイルやログファイルをテキストベースで設定・解析できます)。ファイル管理はプログラム側が行います。デフォルト設定を利用する場合は、本プログラムを立ち上げて数秒でドッキングシミュレーションが開始できます。プログラムが使用するすべのパラメータが完全GUIベースで自由に設定でき、これらパラメータがバーチャルスクリーニング用に開発した様々な高速化手法のパラメータとともにドッキングシミュレーションの流れで配置されています。このため、初心者でも最先端の創薬研究が導入したその日から抵抗なく実施できます(付属マニュアルは実践的なチュートリアルをもちいて全ての機能を紹介しています)。
比類なき、最先端技術
三次元格子点(グリッド)アルゴリズムを用いていないため、急激に落ち込んだポテンシャルエネルギーなどに起因する重要な相互作用エネルギーを見逃すこともなく、また、グリッドアルゴリズムを採用するプログラムで実施されるような結合部位の一部の制限のある構造とのエネルギー計算ではなく、生体高分子システムに含まれる全ての分子、原子を対象に、何らエネルギー計算を簡略化しません。こうした優位性はPIEFII技術により可能となりました。結果的に、計算精度を犠牲にしたりあるいは近似したりしないことから、極めて精度良く安定複合体構造を得ることができます。また、三次元格子点アルゴリズムに特有のプローブ原子等を用いた前処理等も必要なく、これに伴う生体高分子側の制限等も全くありません。グリッドアルゴリズムに対する実際の優位性については後半の図およびFAQで具体的に示します。
ドッキングに使用できる力場として、HyperChemの高信頼性バックエンド計算エンジンが搭載するUnited AtomおよびAll Atom条件下のMM+(MM2改良力場)、Ambers、Amber2、Amber3、Amber94、Amber96、Amber99、OPLS、BIO+83、BIO+85、CHARMM19、CHARMM22、CHARMM27をサポートしており、ドッキングスタディープログラムの中でもこれほどの力場をサポートするものは本プログラムのみです。また、United AtomとAll Atom条件を同時に使用することができるだけでなく、これら条件の境界をボタン操作だけで様々に変更可能です。
ドッキングに用いる化合物の各コンフォメーション毎(構造最適化後)に任意の半経験分子軌道法(CNDO、INDO、NMDO、MINDO3、NMDO/d、PM3、AM1)、により原子電荷がアサインでき、この原子電荷でドッキング後の一連の構造最適化を実施することができます。この機能により、既存システムで唯一、構造変化に伴う電子の再配置を考慮したドッキングシミュレーションが可能です。
その他、HyperChemに搭載されているすべての極小化アルゴリズム(Steepest Descent、Flether-Reeves、Polak-Ribiere、Newton-Raphson)、カットオフや1-4スケールファクター等の力場計算セッティング、シミュレーションにおける各ステップの構造最適化計算の最大サイクルやRMS勾配の閾値設定等もすべて完全GUIベースで自由に変更可能です。距離依存もしくは距離非依存の誘電率を利用して様々な溶媒和条件から真空条件でドッキングシミュレーションが実施できるため、基質結合部位が生体高分子表面上(溶媒接触表面)にある場合でも生体高分子内部(キャビティー)にある場合でも安定複合体構造を高精度に相互作用エネルギー評価できます。さらに、Homology Modeling Professional for HyperChemのGaussian InterfaceおよびONIOM Interfaceと組み合わせることでQM:MM的なドッキングあるいは高レベルab inito量子化学計算を用いた複合体解析(力場計算では取り扱えない結合生成や遷移状態解析あるいは励起状態解析)が可能となり、他に類をみない先進技術が利用できます。
生体高分子側のフレキシビリティはリガンド結合部位周辺残基や水素原子のみに適用することも、あるいは生体高分子システムを構成する水分子、低分子や他の生体高分子などの所望の部分構造または全体構造に適用でき、シミュレーション直前に選択ツールにて画面上の構造を選択するだけで設定が完了します。
生体高分子側はプロテインデータバンクから提供されるデータを編集することなく、そのままドッキングに利用することも可能です。また、複数分子にまたがる結合サイトでも極普通にドッキングシミュレーションできます。さらに、金属原子、低分子、他の生体高分子から成る複雑な分子システムとのドッキングでもHomology Modeling Professional for HyperChemの分子エディタモジュールプログラムを介することで高精度に可能となります。もちろん、ドッキングシミュレーションは生体高分子モデリングあるいは生体高分子結晶構造の精密化とシームレスに並行して実施できるため、リガンド結合部位構造を微調整、検証しながら、ドッキングシミュレーションを実施することも極めて容易です。
相互作用エネルギーの各エネルギー項と活性データの重回帰分析を通して、相互作用エネルギー評価をGUIベースで自由にスコアー化することができます。
リスタート機能をサポートしており、計算精度を損なうことなく、計算途中から再スタートあるいは途中構造からドッキングシミュレーションを開始できます。この機能を利用することでドッキングシミュレーションをバッチ化することが可能となります。トーション数が10を超える非常にフレキシブルな化合物との精密なドッキングシミュレーションを必要とする場合などに有効です。
次世代創薬支援技術、PIEFII技術により、非三次元格子点アルゴリズムで一過程中に複数回の構造最適化を実施しているにもかかわらず、シミュレーションは高速です。簡易スクリーニングモード(ヒット後は構造最適化を複数回実施)のデフォルト条件では、化合物ライブラリにもよりますが、標準的なサイズの生体高分子に対して1CPUあたり1時間で数千化合物をスクリーニング処理する能力があります。*
*1万化合物をスクリーニングするにはUltimate版もしくは簡易スクリーニングオプションを利用する必要があります。
本プログラムはドッキングシミュレーションの正確さを追求したアルゴリズムとなっていますが、バーチャルスクリーニング用に開発した様々な高速化手法(例えば、試行化合物のコンフォメーション対策ではルーズコンフォメーション探索機能やコンフォメーション安定性テスト機能など)も同時に利用可能となっています。
1タスクあたり最大1万化合物までのin silicoスクリーニングに必要な全ての機能を搭載したマルチ化合物対応版も利用できます。また、スクリーニングオプションを追加することで実質、化合物数に制限のない簡易スクリーニングが実施可能です。
上記の先進技術に加え、世界初、ab initioで生体高分子−リガンド結合部位、リガンドのファーマコフォア、およびリガンドのスキャッフォルドまでも高精度に予測する究極の創薬支援技術、PIEFII技術を搭載しています。
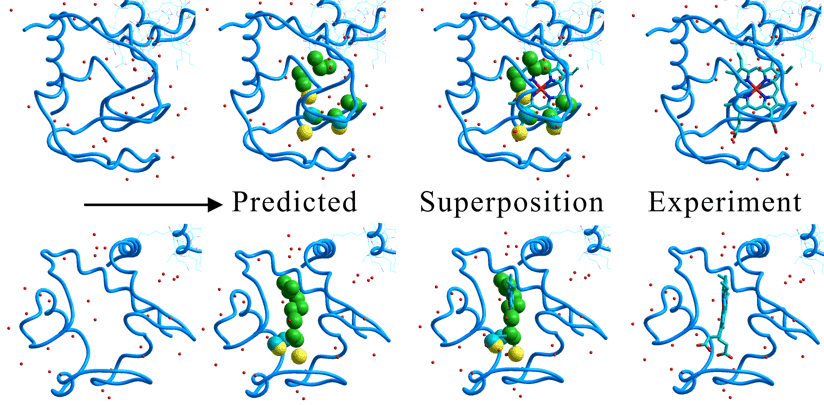
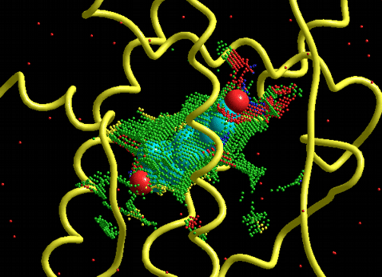
下図はヘムタンパクのチトクロムCからプロトポルフィリンを除去した後に、該タンパクに対してDocking Study with HyperChemに搭載されるPIEFIIプログラムを実施した結果です(上面図と側面図)。左から2個目の図が予測された重原子位置です。予測重原子位置と実際の複合体構造の重ね合わせから(右から2個目の図)、プロトポルフィリンの結合位置のみならずプロトポルフィリンのスキャフォールドならびにファーマコフォアが見事に再現されているのが分かります。さらに、プロトポルフィリンにおけるカルボキシル基の位置やそのコンフォメーションまでも正確に再現しています。仮に本タンパクの補酵素が未知であった場合にでも、PIEFIIによる予測の上面図と側面図からそれがポルフィリンであることは容易にわかります。PIEFII技術に関しては詳細ページを参照してください。
本プログラムを用いたドラッグデザイン方法論および論理的リガンド設計のページが利用可能です。
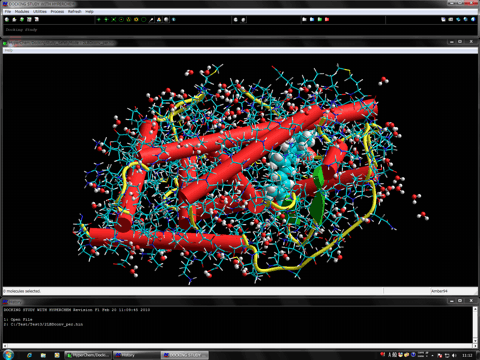
下図はタンパク・リガンドフレキシブルドッキングを用いた天然リガンドの再ドッキングの結果です。化合物については完全なコンフォメーションサーチを実施し、タンパクシステムについてはリガンド結合部位の一部の残基をフレキシブルに取り扱っています。映像中、実際の結晶構造におけるリガンドを赤色のボール&スティックで、ドッキングに使用した化合物を緑色のボール&スティックで示しています。また、フレキシブルに取り扱った残基をチューブで表示しており、レセプターの誘導適合、インデュースドフィット効果がシミュレーションに反映されていることが確認できます(ドッキングシミュレーションで得られた個々のレセプターの座標はあらかじめ重ね合わせてあります)。レセプターには水分子やコファクターも含め全系を対象とした精密なドッキングシミュレーションを実施しています。
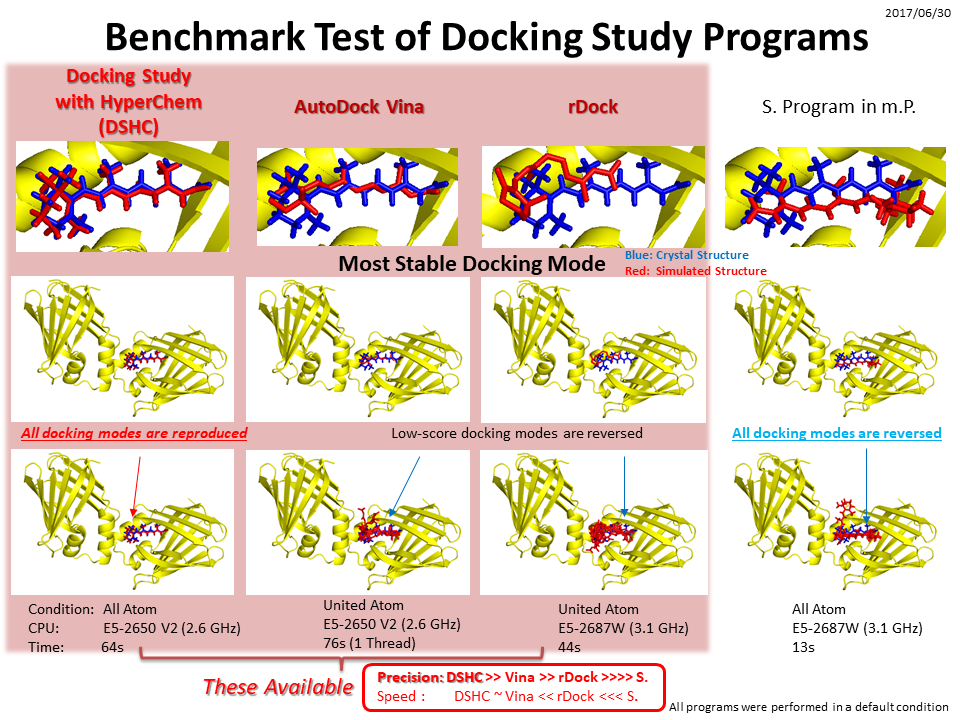
下図はDocking Study with HyperChem(左)とグリッドアルゴリズムを採用とする既存システム(右)との比較です。いずれもデフォルト設定での結果であり、スコアではなく相互作用エネルギーそのものを比較しているのためにプログラムの性能差が明確に現れます。ヒット数、正解率、構造活性相関係数いずれもにおいてもDocking Study with HyperChemが圧倒的な優位性を示しています。
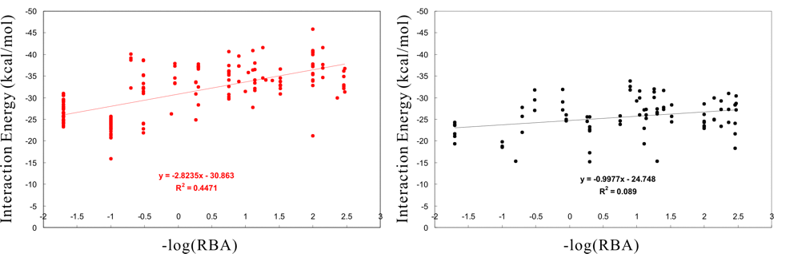
参考データ
Journal of Computer-Aided Molecular Design, 14, 559-572, 2000.
バーチャルスクリーニングプログラムを含む現行のドッキングプログラムの大半はグリッドアルゴリズムを採用しています。グリッドアルゴリズムではドッキングに先立って標的生体高分子(タンパク質、核酸)のリガンド結合部位をグリッドで分割し、いくつかのプローブ原子または分子で格子点上だけを走査し、各格子点またはセルの中心に代表的なエネルギー値を記録しておきます。その後、ドッキングシミュレーションは試行化合物と標的生体高分子(タンパク質や核酸)との間で実施するのではなく、試行化合物と生体高分子の一部の構造に関するグリッドの間で実施されます。そのため、パラメータのないアミノ酸やヌクレオチド以外の分子や金属原子が結合部位にある場合にはこれら分子や金属原子からの立体・電子効果を取り扱えません。最近まで、グリッドアルゴリズムは無限に近いコンフォメーションとドッキングモードを処理するための唯一のテクニックでした。すなわち、実際のドッキングでは試行化合物がオーバーラップしているグリッドに記録されているエネルギー値の総和を計算するだけでよいからです。逆にいうと、グリッドアルゴリズムでは長距離相互作用はもちろん、近距離の相互作用も大幅に近似しており、標的生体高分子(タンパク質分子や核酸分子)の一部の構造以外からの全ての立体・電子効果も明示的に取り扱いません。そのために、しばしば相互作用エネルギーを過小評価し、貧弱な結果をもたらします。また、グリッドアルゴリズムでは通常インデュースドフィット効果など生体高分子側のフレキシビリティーも取り扱えません。
これに対して、Docking Study with HyperChemは次世代創薬支援技術であるPIEFII技術によって、グリッドアルゴリズムを採用することなく、ドッキングシミュレーションを高速に処理できるようになっています。そのため、通常の力場計算条件下に、試行化合物と全体構造の生体高分子(タンパク質分子や核酸分子)との間のエネルギー計算、および生体高分子システムに含まれるその他全ての分子や金属原子とのエネルギー計算を何ら無視することなく明示的に実施しています。さらに、グリッドを利用しないため、生体高分子側をフレキシブルに取り扱えます。その他にも、よりドッキングシミュレーションの精度を重視した独自のアルゴリズムを採用しています。
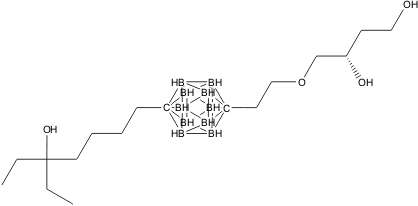
超原子価分子のシミュレーションにも対応
以下は超原子価分子(カルボラン誘導体)の再ドッキングの結果です。青が結晶構造、赤が最安定ドッキングモードを示します。
誘導適合効果を超えた大きな構造変化を取り扱える新たなフレキシブルドッキングアルゴリズムを搭載
既存のスタティックおよび誘導適合効果を考慮したフレキシブルドッキングアルゴリズムと併用して利用できます。なお、本アルゴリズムはアポ体でホロ体のドッキングモードを再現させるという複数の事例で検証済みです。
誘導適合効果を超えた構造変化を取り扱えるフレキシブルドッキングアルゴリズムで探索済みの化合物データベースを再探索することにより、新規骨格を有する高活性なリードもしくは医薬候補化合物がヒットしてくる可能性があります。
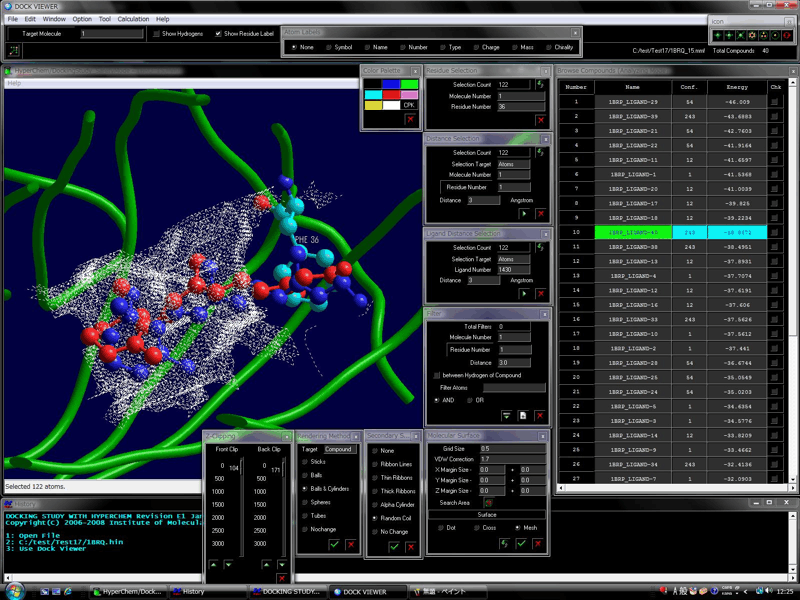
下図は内部構造(フェニルアラニン残基)によって完全に潰された(占有された)アポ体の潜在的な基質結合部位に対して全自動、全く主観なしにタンパク・化合物フレキシブルドッキングシミュレーションを実施した結果(赤色のボール&スティック)です。比較のため、結晶構造解析されたホロ体中での基質(青色のボール&スティック)を重ね合わせています。なお、ホロ体ではフェニルアラニン残基並びにその周囲の主鎖構造は全く異なった位置に移動します(タンパクに関する詳細)。
現行ではペプチドのコンフォメーション探索は低分子と同じ方法のみとなり、現実的にはAutomatic Conformation Search法を利用するか、複数の安定コンフォメーションの初期構造に対して個別に実施するか、特定のねじれ結合を指定したコンフォメーション探索を行うことになります。
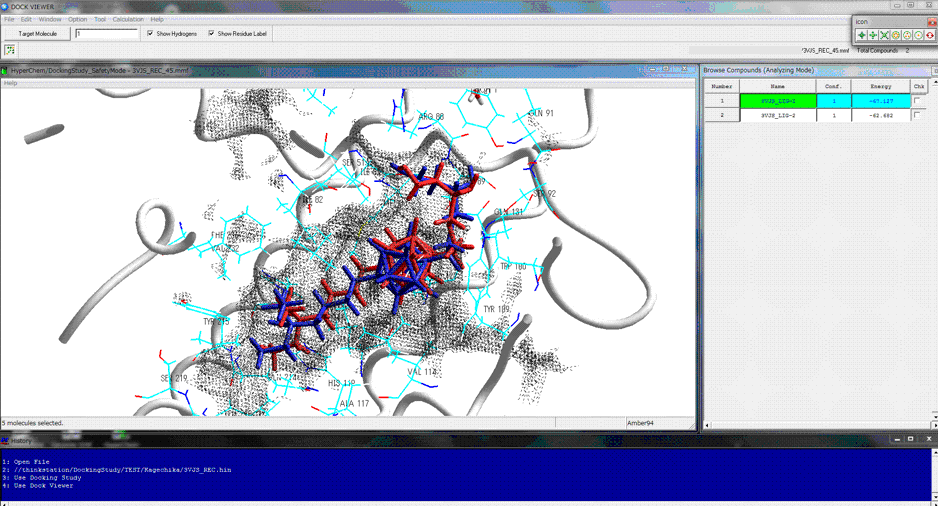
下図はH鎖とL鎖から成る抗体分子とアミノ酸8残基から成るペプチドとの複合体構造についての再ドッキングの結果です(左は全体図、右は拡大図)。ペプチド認識部位はL鎖とH鎖にまたがっています。赤色のボール&スティックがシミュレーションから得られた相互作用エネルギー最安定の複合体構造、青色のボール&スティックが結晶構造解析された実験結果で、両複合体構造はほぼ一致しています。また、本検証ではペプチド認識部位の残基(CPKカラーのボール&スティック)およびその周辺の水分子(赤色のボール)をフレキシブルに取り扱っています。
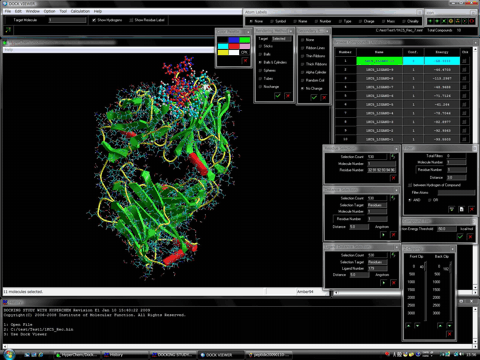
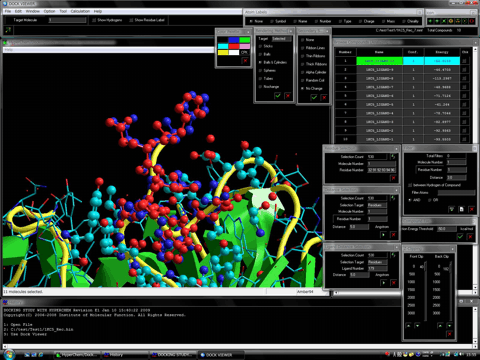
核酸(DNA、RNA)フレキシブルドッキングに対応
下図はDNAとマイナーグルーブバインダーとの再ドッキングの結果です。紫色のボール&スティックが結晶構造、緑色が最安定複合体構造です。なお、ドッキングに使用した核酸分子システムはHomology Modeling Professional for HyperChemで精密化しています。
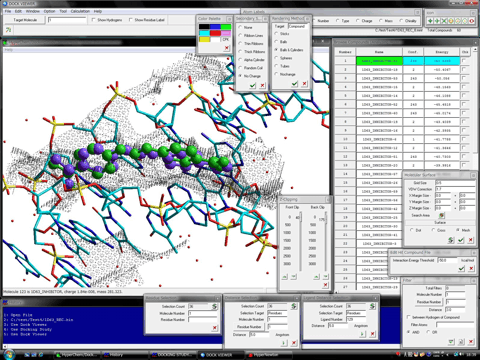
その他の先進機能
生体高分子−リガンド複合体形成部位、リガンドのファーマコフォア、およびリガンドのスキャッフォルドまでも高精度に予測する究極の創薬支援技術PIEFIIを搭載しています。
得られた複合体構造の閲覧には洗練された専用多機能ブラウザー、Dock Viewer、が利用できます。
ドッキングに利用する化合物の初期構造は付属のMol Dimensionが自動的に準備します。もちろん、HyperChemで直接描画して用意した化合物や、X線結晶構造解析などの他の手法で用意した化合物の構造も利用できます。なお、ドッキングに利用する化合物のサイズに制限はありません。
Mol Dimensionが用意した全試行化合物の3D構造は専用の分子ビューア、Mol Browser、で閲覧することができます。
ドッキングに必要なトーションや環構造などあらゆる化合物情報はDocking Studyモジュールプログラムが自動的にアサインします。また、用意されているインターフェイスを利用することで、評価したいトーションだけを利用したコンフォメーション探索や、各トーションに個別の回転角を利用したコンフォメーション探索などの設定も瞬時に準備できます。
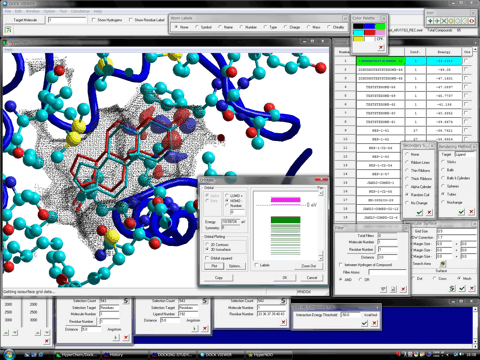
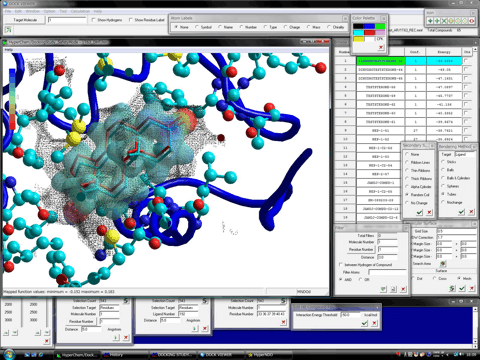
分子の表示はHyperChemバージョン7.5以降であればHyperChemのOpenGL機能をフル活用できます。複合体構造を閲覧しながらデモンストレーションや文献のフィギュアレベルの美しさで自由にそして完全自動でレンダリングされます。リガンド結合部位などの複合体形成部位に分子表面(ファンデルワールス表面)を表示しながら、ヒット化合物の閲覧作業ができます(左図はメッシュ表示、右図はドット表示)。
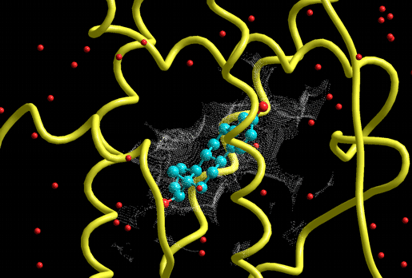
安定複合体を採用したヒット化合物の分子軌道、電荷密度や静電ポテンシャルなどの電子状態を様々な表示形式でレンダリングしながら閲覧作業ができます。左図では最安定複合体を形成したヒット化合物のHOMOを表示し、同時に天然リガンド(赤色のチューブ)と結合部位(キャビティー)の分子表面(黒のメッシュ)も表示しています。右図ではHOMOの代わりに静電ポテンシャルを半透明で同時に表示しています。なお、別に、ヒット化合物と標的タンパク質分子との複合体構造に対する高レベル量子化学計算を用いた詳細な解析はHomology Modeling Professional for HyperChemに搭載されるONIOM Interface for Receptorで可能となります。
導入実績
国内企業(医薬品、食料品、電気機器)、国内独立行政法人研究機関、国内大学研究機関(医学部、薬学部、農学部、工学部、情報科学部)
最新パンフレット (PDFファイル:1MB) 旧パンフレットと併せてご利用ください
リビジョンG1パンフレット (PDFファイル:2MB) 旧パンフレットと併せてご利用ください
パンフレット(PDFファイル:5MB) 旧パンフレットです(ライセンス方式、PIEFII制限原子数が以前の内容となっていますのでご注意ください)
SBDDソリューション資料(PDFファイル:2MB)
製品紹介論文
Motonori Tsuji*. Molecular Science, 1, NP004, 2007.
成果学会報告
成果論文
J. Clin. Immun. 45, 2025.
Biochem. Biophys. Rep., 42, 101981, 2025.
J. Clin. Invest., 135, 2025.
Int. J. Mol. Sci., 25, 13496, 2024.
Microbiol. Spectrum. 12, 2024.
J. Autoimmun. 139, 103085, 2023.
Motonori Tsuji*. Int. J. Mol. Sci. 23, 11009, 2022.
PLOS ONE 16, e0257705, 2021.
Chem. Pharm. Bull. 68, 1193-1200, 2020.
J. Virol. 2020.
Motonori Tsuji*. FEBS Open Bio., 10, 995-1004, 2020.
J. Pharmacol. Exp. Ther. 372, 277-284, 2020.
Motonori Tsuji*. J. Comput. Aided Mol. Des., 31, 577-585, 2017, DOI: 10.1007/s10822-017-0025-6.
Motonori Tsuji*, Koichi Shudo, Hiroyuki Kagechika. FEBS Open Bio., 7, 391-396, 2017, DOI: 10.1002/2211-5463.12188.
Motonori Tsuji*. J. Mol. Graph. Model., 62, 262-275, 2015
Motonori Tsuji*, Koichi Shudo, Hiroyuki Kagechika. J. Comput. Aided Mol. Des., 29, 975-988, 2015.
Motonori Tsuji*. J. Struct. Biol, 185, 355-365, 2014.
Chem-Bio Informatics Journal, 12, 1-13, 2012.
Plant Cell Physiol. 2012 Jul; 53(9): 1638-1647.
Biochemistry. 2010 Dec; 49(50): 10647-10655.
Biochemical and Biophysical Research Communications. 2010 Apr 30; 395(2): 173-177.
Science. 2008 Feb 1; 319(5863): 624-7.
世界が認める最強の分子モデリング環境
HyperChem front-ended
モジュールプログラム
AutoDock Vina In Silico Screenings Interface
| グレード | PIEFII | Docking Study | Dock Viewer | Mol Dimension | Mol Browser | AutoDock Vina Screening*** |
| Essential | 制限版* | 単一化合物 | 無制限 | 単一化合物 | 無制限** | 無制限 |
| Premium Essential | 制限版* | 最大10化合物 | 無制限 | 最大10化合物 | 無制限** | 無制限 |
| Professional | 制限版* | 最大100化合物 | 無制限 | 最大100化合物 | 無制限** | 無制限 |
| Advanced | 制限版* | 最大1,000化合物 | 無制限 | 最大1,000化合物 | 無制限** | 無制限 |
| Ultimate | 制限版* | 最大10,000化合物 | 無制限 | 最大10,000化合物 | 無制限** | 無制限 |
|
Cluster Virtual Screening System |
非制限版 | 無制限、クラスター | 無制限 | 無制限、クラスター | 無制限 | 無制限 |
*最大同時予測可能原子数1000原子、約80残基
**プログラム自体に制限はありませんが、実際に利用できる数はDocking StudyまたはMol Dimensionモジュールプログラムに依存します。
***グレード(スクリーニング能力)に関係なく、AutoDock Vinaスクリーニングには化合物数による制限はありません。
追加オプション*
Docking Studyモジュール: 簡易スクリーニングオプション(化合物数無制限)
Mol Dimensionモジュール: 化合物数変更オプション
*分散処理には対応していません。分散処理を利用するにはVirtual Screening Systemが必要になります。
推奨最小システム構成
プロセッサー:マルチコアIntel CoreまたはXeon 2GHz以上(Intel Pentium III以降)
オペレーティングシステム:Microsoft Windows 10及び11(Microsoft Windows 98以降、全機能を利用するにはXP以降が必要)
メモリー (RAM):8 GB以上(最低256 MB以上)
モニタ:SXGA以上(フルHD以上を推奨)
グラフィックスボード:OpenGL対応ボードを推奨
マウス:キーストロークを割り当てられる多機能マウスを推奨
その他:HD 512GB(巨大(1億化合物)SDFまたはMOL2ファイルを取り扱う場合は2TB)以上を推奨; CD-ROMドライブ、ネットワークカード
必要なソフトウェア
必須
HyperChem(Windows版;Student Versionは除く):HyperChem Release 8.0.10 Professional for Windows (Release 6.0.3以降、全機能を利用するにはRelease 7.5.2以降が必要)
TclPro1.2(Windows版):Docking Study with HyperChemの使用にはTclPro1.2(Windows版)が必要です。TclPro1.2はウェブ上から無料で入手できます。
オプション
AutoDock Vina(Windows版またはUnix版):AutoDock Vinaでインシリコスクリーニングまたはドッキングシミュレーションを実施する場合に必要*
*AutoDock VinaによるインシリコスクリーニングおよびドッキングシミュレーションはDocking Study with HyperChemとは全く異なるプログラムであることに注意してください。Docking Study with HyperChemはその優れたGUI環境でAutoDock Vinaインシリコスクリーニングのためのインターフェイスを提供しているだけです。AutoDock Vinaスクリーニングを実施するにあたり、パッケージグレード(スクリーニング能力)による化合物数の制限はありません。
OpenBabel(Windows版、64ビット版または32ビット版):AutoDock Vinaインシリコスクリーニングを実施する場合およびHIN形式(HyperChemフォーマット)またはPDB形式からMDL SDF形式、Tripos MOL2あるいはAutoDock PDBQT形式で出力する場合に必要
連携
rDock、SMINA、GNINA(Unix版):rDockやSMINA・GNINAドッキングプログラムで使用する入出力ファイルを取り扱えるため、rDockやSMINA・GNINAの入力ファイル作成、化合物三次元ライブラリ作成、及び結果ファイルを閲覧、解析、ヒット絞り込みできます。*
*rDockやSMINA・GNINAはDocking Study with HyperChemとは全く異なるプログラムであることに注意してください。Docking Study with HyperChemはその優れたGUI環境でrDockやSMINA・GNINAでのドッキングシミュレーション結果を閲覧、解析、ヒット絞り込みするためのインターフェイスを提供しているだけです。
その他、ナノ秒スケールの分子動力学計算を実施する場合はNAMDおよびVMD、全系量子力学計算を実施する場合はABINIT-MPおよびBioStation Viewer、GAMESSおよびFuまたはFacioを各ソフトウエアのライセンスに基づいて各自の責任において用意してください。
備考
PDBフォーマットバージョン2.3、3.0-3.3対応
Docking Study with HyperChemはTcl/Tkプログラム(主にインターフェイス)と32ビットWindowsアプリケーションからなるソフトウェアパッケージです。
本ソフトウェアパッケージはTclPro1.2コンパイラーでコンパイルされています。そのため、ソフトウェアパッケージの使用には、パッケージのインストールに先立って、TclPro1.2のインストールが必要です。
充実したオンラインヘルプがソフトウェアパッケージの個々のプログラムから使用できます(英文)。
全機能を実践チュートリアル形式で紹介した日本語版ユーザーマニュアル第1巻と第2巻(同種既存システムのマニュアルはもちろん、文献、書籍からも絶対に得られない最先端ノウハウを満載し、文章だけで108ページおよび54ページにわたる詳細解説)が付属します。
リモートデスクトップ及びマルチコア環境
Docking Study with HyperChemはCPUコア数分を同時に実行できます。このため、1台のPCワークステーションでも分散処理できます。
最上位グレードのCluster版はリモートデスクトップ機能を利用して複数台のPCクラスターで化合物データベースを全自動で分散処理する機能が用意されています。
最上位グレードCluster版とHyperChemサイトライセンス版を複数PCワークステーションに導入することによって本格的なバーチャルスクリーニングが可能となります。本製品(バーチャルスクリーニングシステム)は個別対応とさせていただいています。詳しくは下記リンクをご確認ください。
重要:本ソフトウェアパッケージをインストールするにあたり、まずシステムにHyperChemとTclPro1.2がインストールされていることを確認してください。このパッケージにはHyperChemは含まれていません。
*HyperChemはHypercube, Incの登録商標です。
**本製品の開発にあたり、Hypercube, Inc.の承認済みです。