![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
HyperChem取り扱い
分子モデリングソフト、分子シミュレーションソフト、計算化学ソフト:費用対効果に優れたデファクトスタンダードの高機能・高性能な統合分子設計支援システム
HyperChem
Release 8
長年、世界中の化学者に愛用されている費用対効果に大変優れたデファクトスタンダードの高機能&高性能な分子モデリング・分子シミュレーション・計算化学ソフト。
あらゆる分子種を表現可能な平面分子作画機能、自動三次元化機能、自動水素付加機能、自動力場設定機能、アミノ酸・核酸・糖・ポリマーエディタ・クリスタルビルディング機能など卓越した分子モデリング機能。各種の分子力学計算、半経験分子軌道法、量子力学計算、密度汎関数法、分子動力学計算、極小化アルゴリズムが利用できる網羅的計算化学環境。豊富なレンダリング機能、ユーザーによる機能拡張性、各種ファイルフォーマットに対応した互換性などが特徴。




プライベートアドレスクラスC(256台)などのサイトライセンスもご利用いただけます
高校、高等専門学校、大学、大学院での化学教育の教員・学生の方はこちら
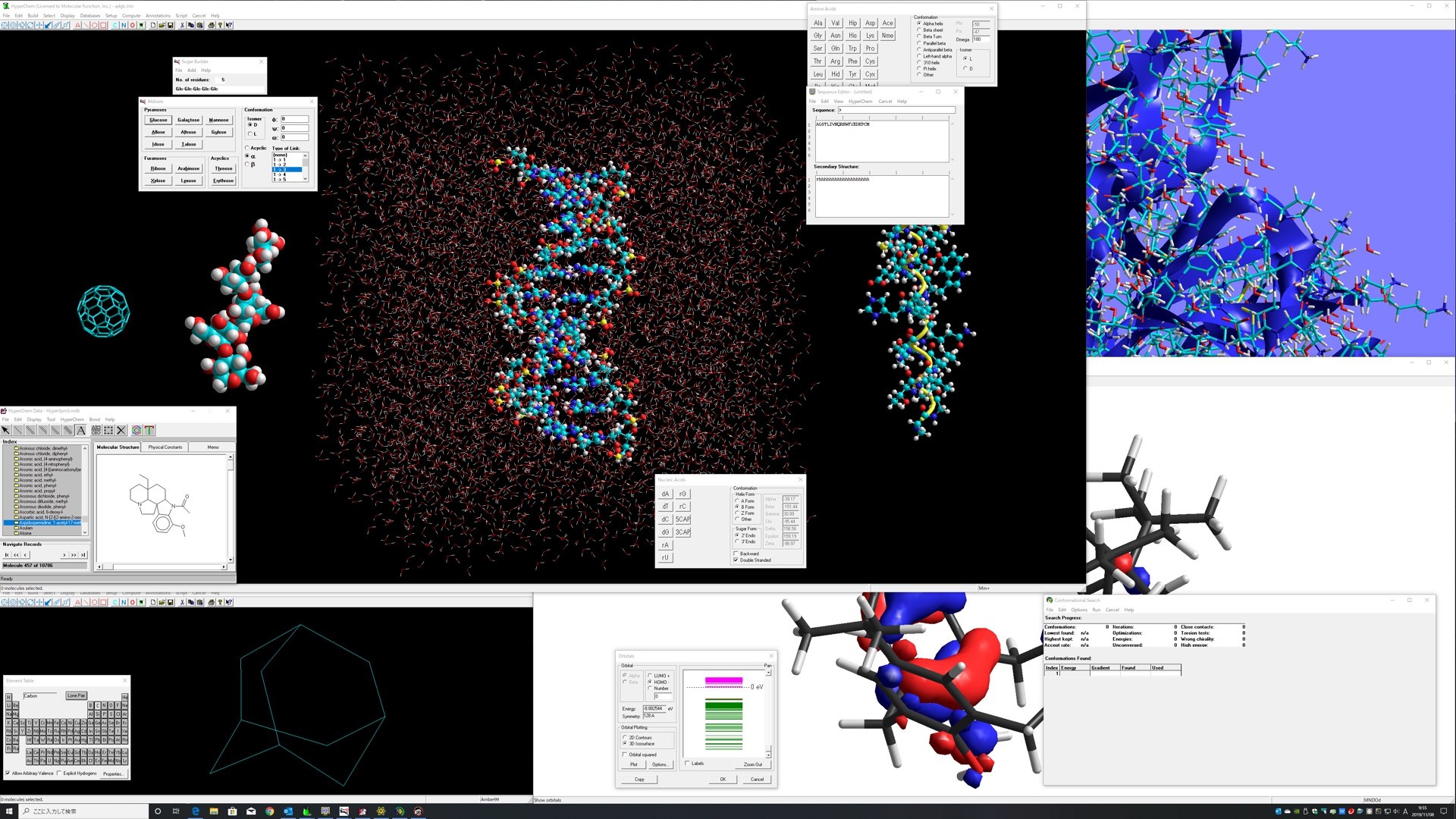
HyperChem機能 構造式ドロー・分子モデリング・分子シミュレーション・量子化学計算シミュレーション・分子動力学(分子力学から量子化学まで可能)シミュレーション・10万原子を超える複雑系システムの分子モデリング・スクリプタブル
- 洗練された分子モデリング環境 -
卓越した分子モデリング機能
あらゆる分子種を表現可能な平面分子作画システム(10万原子以上表示可能)
自動三次元化、自動水素原子付加、自動力場パラメータ設定機能
アミノ酸、核酸、糖、ポリマーエディタ、クリスタルビルダー機能
RMSフィット、オーバーレイ機能
コンフォメーション探索機能
カット&ペースト機能
対称性操作、リストレイン・コンストレイン設定機能
細かな選択機能と選択部分のみでの部分計算機能やレンダリング機能
網羅的計算化学環境
分子力学計算(MM)
MM+、Ambers、Amber2、Amber3、Amber94、Amber96、Amber99、Bio+83、Bio+85、Charmm-19、Charmm-22、Charmm-27、Opls、Custom
量子化学計算・分子軌道法・拡張ヒュッケル法
半経験分子軌道法(QM)
Extended Huckel、CNDO、INDO、MINDO3、MNDO、MNDO/d、AM1、PM3、RM1、ZINDO/1、ZINDO/s、TNDO
Ab Initio分子軌道法 (QM)
(QM)
Hartree-Fock、MP2、CI
密度汎関数法(QM)
一点計算、構造最適化
分子動力学計算(量子化学計算QMと分子力学計算MMの両方に対応)
Molecular Dynamics、Langevin Dynamics、Monte Carlo
極小化アルゴリズム
Steepest Descents、Flecher-Reeves、Polak-Ribiere、Eigenvector Following、Newton-Raphson、Conjugate Directions
振動解析、遷移状態解析、NMRケミカルシフト予測、IRチャート予測、紫外吸収波長予測、励起状態解析、QSAR解析、コンフォメーション探索(量子化学計算QMと分子力学計算MMの両方に対応)
QM/MM計算も可能、周期境界条件自動設定
豊富なレンダリング機能
選択的表示機能、Customカラー表示
ラベル機能
symbol、name、number、type、charge、spin、mass、chirality、basis set、RMS gradient、residue、sequence、res+seq、bond length、bond order、custom
分子表示 
スティック、ボール、ボール&スティック、CPK、チューブ、ドット表示
分子軌道、静電ポテンシャル、電荷密度、スピン密度表示
2D-3D、メッシュ、Jorgensen-Salem、ライン、フラット、半透明、その他
水素結合、双極子モーメント
OpenGL二次構造表示、リボン表示
反応座標、NMRチャート、振動チャート
拡張性
HCL(HyperChem Command Language)スクリプタブル
C、C++、Fortran、Tcl/Tk、DDEスクリプタブル(Windows版のみ)
互換性
PDB(ENT)、カーテシアン、Z-マトリックス、ISIS、MDL、TRIPOS、ChemDraw
サードパーティー製計算化学プログラムインターフェイス(Windows版のみ)
Gaussian、Q-Chem、GAMESS、Firefly、PQS、MOPAC2009
推奨最小システム構成
プロセッサー: インテルCoreまはたXeon以降 (1GHz以上)
オペレーティングシステム: マイクロソフトWindows 10及び11 (32ビットおよび64ビット (WOW64)版に対応)(Windows95以降に対応)
Mac: Mac OS X, OS X, macOS10.x*
Linux:*
メモリー (RAM): 2GB以上(最低256 MB以上)
ハードディスク:128GB以上(本体のみで300MB必要)
その他: DVD-ROMドライブ、キーボード、マウス(タッチパネルにも対応可能)
*注意: MacおよびLinux版は使用環境に依存します。必ず、ご使用環境で使用可否を評価版にてご確認ください。
インストール手順と初期設定
インストール方法とインストール時の初期設定方法は以下のYouTubeをご覧ください。
備考
Gaussianプログラムなどのサードパーティー製計算化学エンジンとも高度に連携できます。
HyperChemはマルチコア対応ではありませんが、同時に複数スレッドを立ち上げることが可能なため、コア(スレッド)数分の独立したHyperChemが同時に実行できます。また、ワークスペース内の個々の分子を個別に計算対象とできます。
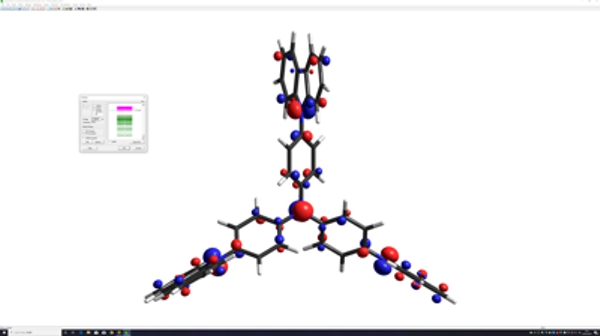
HyperChemなら誰でも反応設計、医薬分子設計から機能性材料設計を手軽に実施できます。

HyperChemなら誰でも簡単に分子力学計算はもちろん量子化学計算でコンフォメーション探索を実施して最安定コンフォメーションを得ることができ、機能材料設計を論理的に支援します。

HyperChemなら誰でも簡単に10万原子を超える巨大生体高分子のモデリング、シミュレーションが行えます。

国内唯一のHyperChem正規ディーラー
米Hypercube社に直接問い合わせはできません。分子機能研究所を必ず経由してください。
ご利用目的に応じて分子機能研究所が万全の態勢でサポートします。
![]()
![]()
HyperChemのパンフレットはこちら
HyperChem評価版(完全版)の入手方法
ダウンロード方法、インストール手順、10日間の評価用ライセンス発行に対応します。
評価版は期間限定の完全版ですので、製品ご購入時に改めてインストールする必要がなく、USBハードウエアロックをパソコンに挿入するだけでそのまま使用できるようになります。
注意:HyperChemのフリーダウンロードサイトにご注意ください。Hypercube社公式サイト以外からのトライアル版のダウンロードは大変危険です。
HyperChem Release 7 完全マニュアル (約20 MB)
HyperChem Release 8 完全マニュアルは製品からPDFファイルで自動インストールされます。
旧HyperChemマニュアルです。Release 8でも基本操作はほぼ同じですが、Release 8から選択と機能が分離されていることに注意が必要です。
HyperChemを用いた各種チュートリアルはYouTube上の動画を参考にしてください。

*HyperChemはHypercube, Incの登録商標です。