![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
ドッキングスタディー、ドッキングシミュレーション、分子ドッキング法
ドッキングシミュレーションアルゴリズム
構造ベースによる化合物の標的生体高分子リガンド結合部位への重ね合わせ手法
重ね合わせは、化合物およびリガンド結合部位におけるある化学的特徴を反映させたフラグあるいはグリッドやいくつかの球で形成される形状に対して実行されます(個々のドッキングシミュレーションプログラムの特徴はこの化学的性質を反映したものとなっています)。
多くのドッキングシミュレーションプログラムでは、生体高分子側(タンパク、核酸等)を静的に扱い、側鎖のインデュースドフィット効果等に関しては力場ポテンシャル等で対応していますが、最近では、リガンド結合部位残基側鎖のフレキシビリティを考慮したフレキシブルドッキングが可能なものもあります。なお、化合物のコンフォメーションだけを考慮したプログラムでもフレキシブルドッキングシミュレーションプログラムと呼ぶため注意が必要です。
一般に、化合物の無数のコンフォメーション対策のために、重ね合わせに成功した後に、化合物の二面角を任意の刻み幅(=精度;30°、45°、60°、90°あるいは120°)で回転させます。続いて、構造最適化計算を用いて二面角回転で得られたモードについて相互作用エネルギーが見積もられます。ただし、本アルゴリズムの場合、シミュレーション時間は短縮されますが、ドッキングシミュレーションの精度を犠牲にします。この問題を回避するために、本アルゴリズムを採用するドッキングシミュレーションプログラムではドッキングに使用する化合物の初期構造を前もって複数用意しておく必要があります。
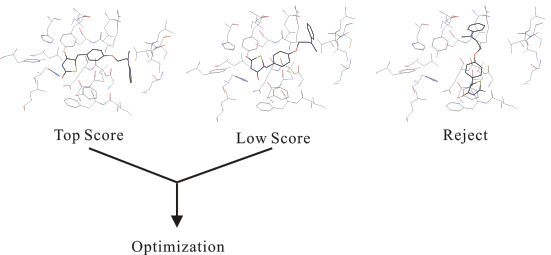
構造最適化の後、個々のドッキングモデルはドッキングシミュレーションプログラムが独自に搭載するエネルギー関数(用いる系にあわせて変数を調整するスコア計算)を用いて評価されます。これは、既存のプログラムの大部分がグリッドアルゴリズムを採用していることと関係があります。グリッドアルゴリズムでは、相互作用エネルギー計算が大幅に簡略化されるためにそのままでは利用できず、扱う系に対してパラメータをチューニングしたスコア計算(あるいは得られた複合体構造に対して別途結合自由エネルギー計算)を用いるのが一般的です。
そのうち、高いスコアーの複合体モデルの化合物が標的生体高分子(タンパク、核酸)に対する活性候補化合物として提示されます。
グリッドアルゴリズム
バーチャルスクリーニングプログラムを含む現行のドッキングシミュレーションプログラムの大半はグリッドアルゴリズムを採用しています。グリッドアルゴリズムではドッキングに先立って標的生体高分子(タンパク質、核酸)のリガンド結合部位をグリッドで分割し、いくつかのプローブ原子または分子で格子点上だけを走査し、各格子点またはセルの中心に代表的なエネルギー値を記録しておきます。その後、ドッキングシミュレーションは試行化合物と標的生体高分子(タンパク質、核酸)との間で実施するのではなく、試行化合物と生体高分子の一部の構造に関するグリッドの間で実施されます。そのため、パラメータのないアミノ酸やヌクレオチド以外の分子や金属原子が結合部位にある場合にはこれら分子や金属からの立体・電子効果を取り扱えません。最近まで、グリッドアルゴリズムは無限に近いコンフォメーションとドッキングモードを処理するための唯一のテクニックでした。すなわち、実際のドッキングシミュレーションでは試行化合物がオーバーラップしているグリッドに記録されているエネルギー値の総和を計算するだけでよいからです。逆にいうと、グリッドアルゴリズムでは長距離相互作用はもちろん、近距離の相互作用も大幅に近似しており、標的生体高分子(タンパク質、核酸)の一部の構造以外からの全ての立体・電子影響も明示的に取り扱いません。そのために、しばしば相互作用エネルギーを過小評価し、貧弱な結果をもたらします。また、グリッドアルゴリズムでは通常インデュースドフィット効果など生体高分子側(タンパク、核酸)のフレキシビリティーも取り扱えません。
バーチャルスクリーニングに戻る
2005年5月
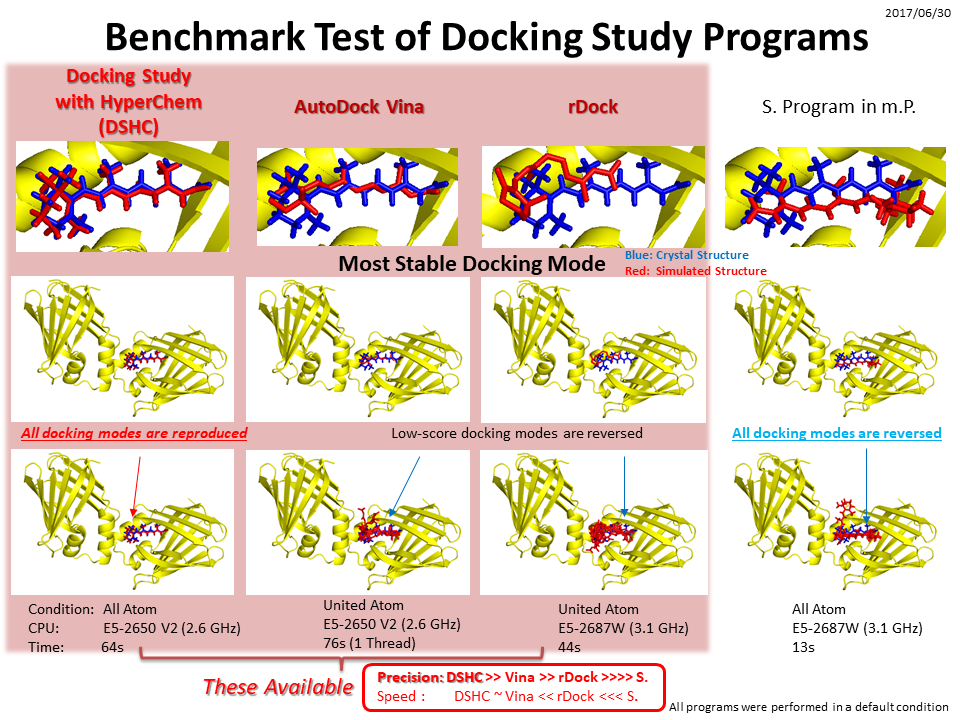
さらに詳細なアルゴリズムについてはDocking Study with HyperChemを参照してください。
Docking Study with HyperChemプログラムはドッキングシミュレーションの正確さを追求したアルゴリズムを採用する次世代創薬支援技術搭載、無制限疑似クラスターでのin silicoスクリーニングが可能な、真の生体高分子・リガンドフレキシブルドッキングシミュレーションプログラムです。
受託研究・共同研究
ドッキングスタディーの受託研究・共同研究を受け付けています。20年以上の豊富な実績があります。お気軽にお問い合わせください。
| MFDDインシリコ創薬受託研究サービス 詳しくはこちら |