

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
Molecular Modeling and Structure-based Drug Design Systems
Gaussian Interface for HyperChem
Computational Chemistry Environment Based on HyperChem GUI
Compatible to Gaussian94, Gaussian98, Gaussian03, Gaussian09, Gaussian16 & GaussView2, GaussView3, GaussView4, GaussView5, GaussView6
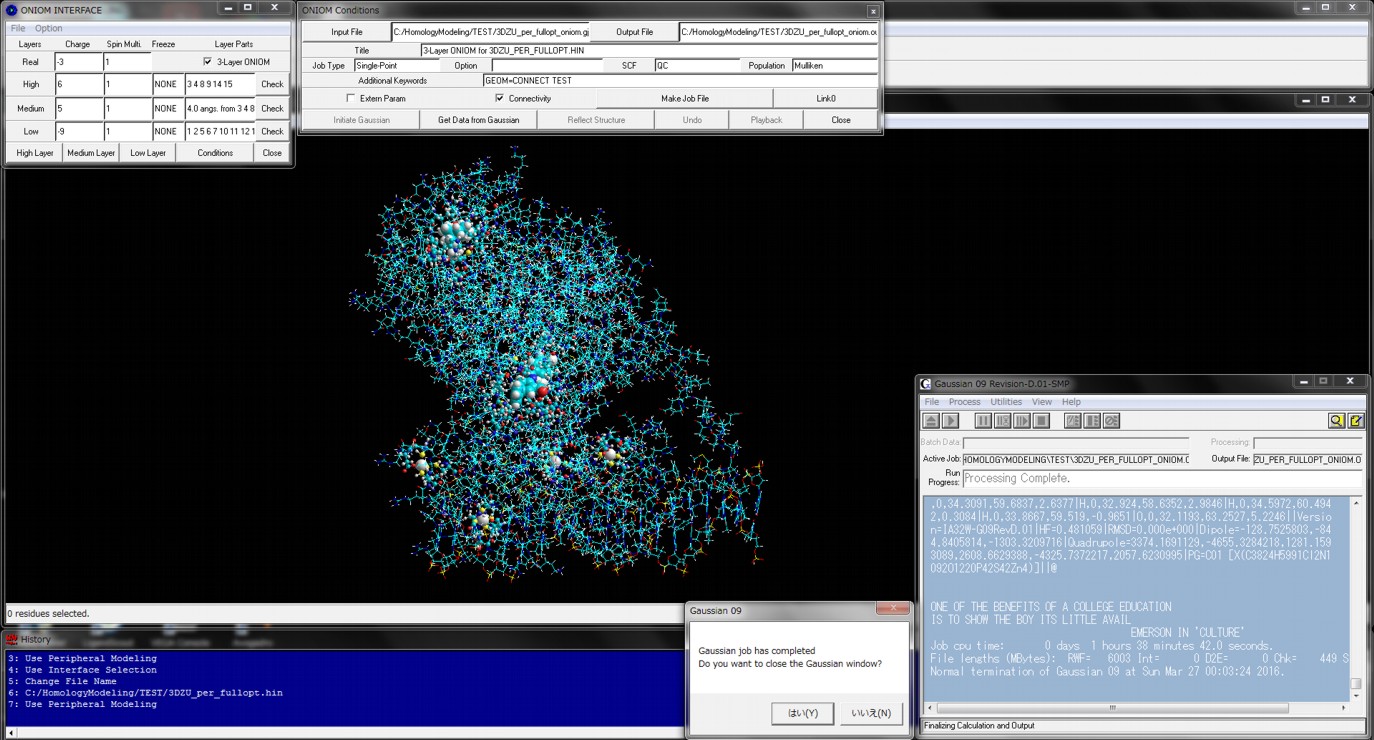
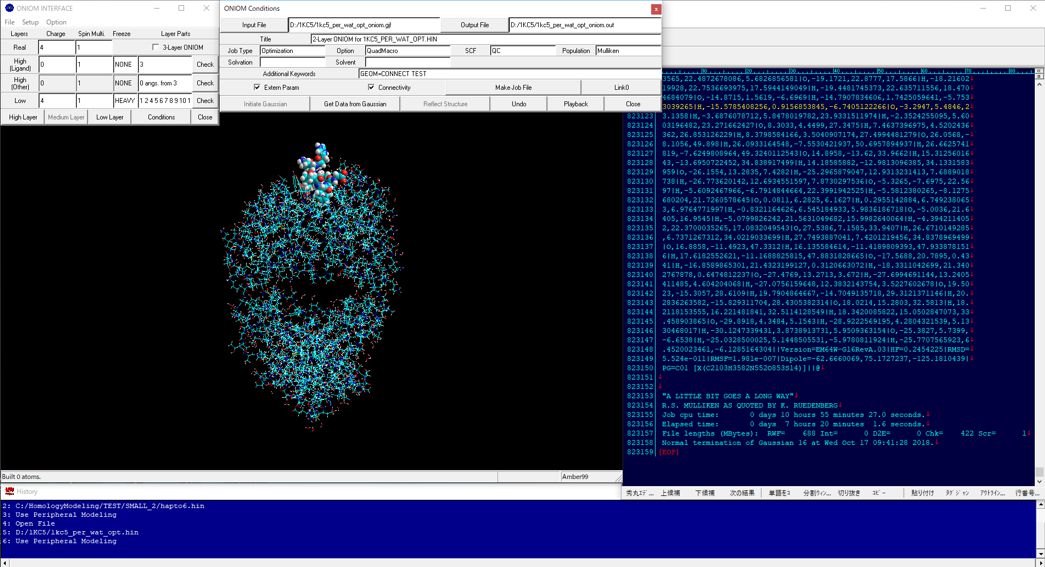
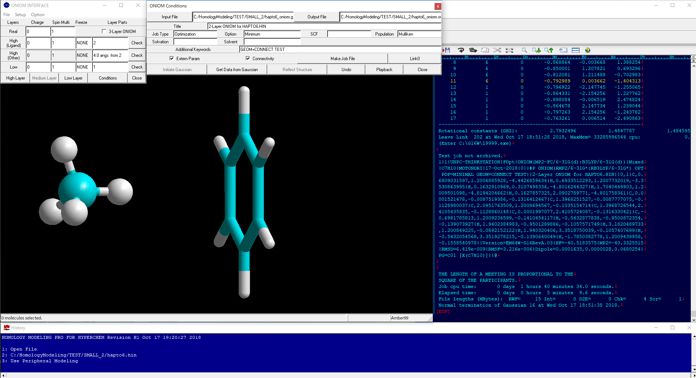
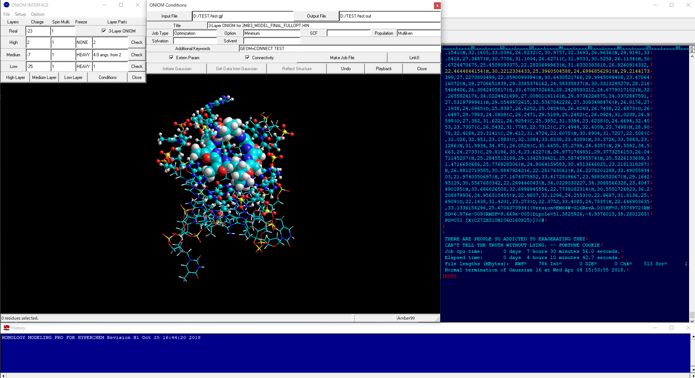
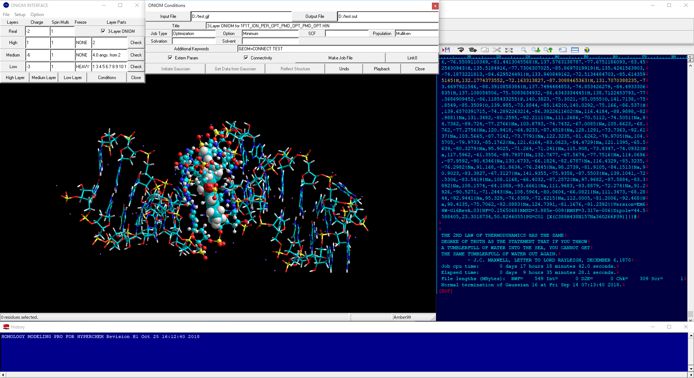
In quantum mechanics calculations for the entire structure of big molecular system such as biomacromolecular system, the single point calculations for the initial structure obtained from classical molecular mechanics calculations and/or classical molecular dynamics calculations cannot converge or will give abnormal energies. For obtaining reliable results from quantum mechanics calculations such as fragment molecular orbital methods of entire molecular system, the initial structure must be prepared using geometrical optimization calculations via ONIOM methods. ONIOM Interface for Receptor can provide the best solution for the quantum mechanics calculations of the entire molecular system of which the precise initial structure is prepared from Docking Study with HyperChem and Homology Modeling Professional for HyperChem.
Motonori Tsuji, et. al. FEBS Open Bio., 7, 391-396, 2017, DOI: 10.1002/2211-5463.12188.
The most powerful and sophisticated molecular modeling environment
HyperChem front-ended
The most powerful computational chemistry environment
Gaussian back-ended
Feature
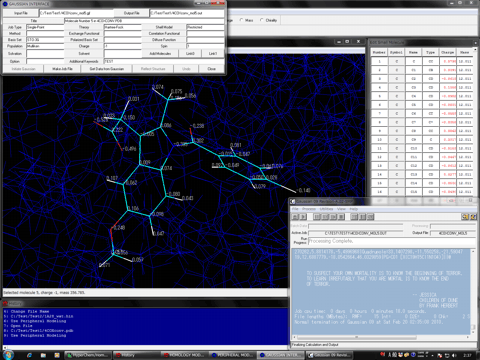
"Gaussian Interface for HyperChem" can automatically prepare a Gaussian input file, "job file", for a molecular system constructed using the powerful molecular modeling function of HyperChem. The calculation results (atomic charges and optimized structure) of the Gaussian job can be immediately reflected to the corresponding molecule in the HyperChem workspace.* The interface can prepare almost all of major methods and options except for the ONIOM method and supports the Link1 calculations of Gaussian. When a multi-step job is used, the interface can reflect these results, separately. When some molecules exist in the HyperChem workspace, the interface can treat these molecules independently or simultaneously.
Gaussian Interface for HyperChem is an add-on program to the Peripheral Modeling program of the Homology Modeling Professional for HyperChem package. Therefore, this program serves as a logical molecular modeling tool for preparing a small molecule in a biological macromolecular system, as well as can be used for obtaining the chemical properties of a simple small molecule. When the interface is used for a biological macromolecular system including some small molecules, the interface can provide a simple, high-speed, and highly precise method for preparing the atomic charges for the entire system instead of the QM/MM method such as the ONIOM method (see the advanced usages and tutorial).
The full-automatic ONIOM Interface to the ONIOM method of Gaussian is also available from the Peripheral Modeling program of the Homology Modeling Professional for HyperChem package.
Additional Functions
Moreover, since HyperChem carries a Z-matrix interface for Mopac5, 6, and 7 in default, their Gaussian jobs can be initiated using Gaussian Interface for HyperChem without any conversions to the corresponding internal coordinate. The latest version is compatible to the fragment molecular orbital calculation programs, ABINIT-MP (BioStation Viewer) and GAMESS (Fu / Facio) and the molecular dynamics calculation program, NAMD (VMD). If Q-Chem for Windows and a set of MOPAC2000 and Mopac Interface for HyperChem are available, the HyperChem - Gaussian - Mopac - Q-Chem - ABINIT-MP - NAMD compatibility becomes possible on the HyperChem graphical user interface, thus giving a powerful computational chemistry environment.
* When a checkpoint file name is specified in the Link0 entry box of Gaussian Interface for HyperChem, the results of the corresponding Gaussian job can be analyzed in detail, using a Gaussian viewer through the checkpoint or formcheck file prepared.
References
Motonori Tsuji, Molecular Science. 1, NP004, 2007.
Science. 319, 624-7, 2008.
Biochemical and Biophysical Research Communications. 339, 173-177, 2010.
Biochemistry. 49, 10647-10655, 2010.
Plant Cell Physiol. 53, 1638-1647, 2012.
Motonori Tsuji, Journal of Structural Biology. 185, 355-365, 2014.
Motonori Tsuji, et. al., Journal of Computer-Aided Molecular Design., 29, 975-988, 2015.
Motonori Tsuji, Journal of Molecular Graphics and Modelling., 62, 262-275, 2015.
Motonori Tsuji, et. al., FEBS Open Bio., 7, 391-396, 2017. DOI: 10.1002/2211-5463.12188.
Motonori Tsuji. Journal of Computer-Aided Molecular Design., 31, 577-585, 2017, DOI: 10.1007/s10822-017-0025-6.
J. Pharmacol. Exp. Ther. 372, 277-284, 2020.
Motonori Tsuji. FEBS Open Bio., 10, 995-1004, 2020.
J. Virol. 2020.
Chem. Pharm. Bull. 68, 1193-1200, 2020.
PLOS ONE 16, e0257705, 2021.
Motonori Tsuji. Int. J. Mol. Sci. 23, 11009, 2022.
Description
Gaussian Interface for HyperChem has been integrated into the Peripheral Modeling Professional module program in Homology Modeling Professional for HyperChem. Refer also to the Peripheral Modeling Professional module program.
New, High-Performance Software for Biomacromolecule Modeling, Functional Analysis, and Simulation.
Homology Modeling Professional for HyperChem
with Gaussian Interface for HyperChem & ONIOM Interface for Receptor
Compatible to NAMD Molecular Dynamics and ABINIT-MP / GAMESS Fragment Molecular Orbital Quantum Mechanics Calculations for Entire Molecular System
* HyperChem is a registered trademark of Hypercube, Inc.
** Gaussian is a registered trademark of Gaussian, Inc.