

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
Molecular Modeling and Structure-based Drug Design Systems
The True Flexible Docking Simulation and In Silico Screening Program Package
Docking Study with HyperChem, Revision H1
Essential, Premium Essential, Professional, Advanced, Ultimate, and Cluster
with AutoDock Vina In Silico Screenings Interface
Compatible to rDock Docking simulations, NAMD Molecular Dynamics and ABINIT-MP / GAMESS Fragment Molecular Orbital Quantum Mechanics Calculations for Entire Molecular System
- Carrying The Ultimate Technology In The Drug Discovery -
The Latest In Silico Drug Design Platform: Evolution of Docking Study with HyperChem, Revision H1.
Supported AutoDock Vina Virtual Screening Interface (preparation of compound database - preparation of all input files - execution - viewing and filtering of hits; supports also simultaneous analysis of both Docking Study and AutoDock Vina results) and rDock docking simulation interface.
In quantum mechanics calculations for the entire structure of big molecular system such as biomacromolecular system, the single point calculations for the initial structure obtained from classical molecular mechanics calculations and/or classical molecular dynamics calculations cannot converge or will give abnormal energies. For obtaining reliable results from quantum mechanics calculations such as fragment molecular orbital methods of entire molecular system, the initial structure must be prepared using geometrical optimization calculations via ONIOM methods. ONIOM Interface for Receptor can provide the best solution for the quantum mechanics calculations of the entire molecular system of which the precise initial structure is prepared from Docking Study with HyperChem and Homology Modeling Professional for HyperChem.
Motonori Tsuji, et. al. FEBS Open Bio., 7, 391-396, 2017, DOI: 10.1002/2211-5463.12188.
The full-automatic and comprehensive biological macromolecule (protein and nucleic acid molecule systems)- and ligand (small molecule and peptide)-flexible docking simulations and in silico screenings program.
It's easy to treat the comprehensive conformation search of compound, induced fit effects of the side chain and main chain of the biological macromolecule, and the flexibility of the other molecules such as water molecules, biological molecules, small molecules, and metal atoms, etc.
The program (non-grid algorithm) can predict the precise interaction energy for the entire system rather than the approximated interaction energy obtained from the other docking simulation programs which adopt the grid algorithm for simplifying the energy calculations.
The program adopts the high reliable search method of stable complex using our novel docking algorithm based on the PIEFII technology which can precisely predict the binding site and ligand pharmacophore points in terms of the structure based manner.
In addition to general functions required for the docking simulations and screenings, the flexible docking function in consideration of the induced fit effect of biological macromolecules, the flexible docking function in consideration of large structural changes such as transformation between apo and holo conformations, the flexible docking function under the steric and electronic influences from any other molecules such as the macromolecules, small molecules, water molecules, metal atoms, and substituent groups bound to the target molecule, the atomic charge reassignment function for each conformations using any semi-empirical molecular orbital methods, any combinations of the united atom and all atom conditions, the restart function, the docking function under the solvated conditions, clustering function can be used for performing the precise docking simulations and in silico screenings. Moreover, power users can freely alter any parameters for the docking simulations and screenings as well as those for the molecular mechanics and quantum mechanics calculations via the provided graphical user interfaces.
You can perform the latest docking simulations and in silico screenings without difficulty.
AutoDock Vina In Silico Screening Interface is now available for docking simulations and virtual screenings using AutoDock Vina program. In addition, all docking algorithms such as biomacromolecule- & ligand flexible docking, ligand flexible docking, rigid docking, and partial structural docking of Docking Study module program can simultaneously carry out.
The protein- and ligand-flexible docking program, Docking Study with HyperChem, is now available. This program is based on the excellent computational chemistry environment of HyperChem.
The full-automatic and comprehensive biological macromolecule (protein and nucleic acid molecule systems)- and ligand (small molecule and peptide)-flexible docking simulation program. It's easy to treat the comprehensive conformation search of compound, induced fit effects of the side chain and main chain of the biological macromolecule, and the flexibility of the other molecules such as water molecules, biological molecules, small molecules, and metal atoms, etc. The program (non-grid algorithm) can predict the precise interaction energy for the entire system rather than the approximated interaction energy obtained from the other docking simulation programs which adopt the grid algorithm for simplifying the energy calculations.
The High-Speed and High-Performance, Protein- and Ligand-Flexible Docking Simulation Program
Docking Study with HyperChem can predict the best docking mode of a complex between protein and compound, and can suggest the directionality of molecular design in the structure-based manner. Therefore, this program can support the high-level drug design such as the lead optimizations as well as can predict the lead compounds.
The program can carry out the fully-automatic, comprehensive docking calculations under contributions of a protein flexibility (main chain and side chain flexibilities), as well as under contributions of a compound flexibility using full conformation search. Thus, the program can obviously deal with the induced fit effect of the protein system.
This program supports the full graphical user interface basis (It is not necessary to edit and prepare a parameter file.).
Advanced Technology
Since the energy calculations do not use the grid algorithm, the program does not underestimate a critical interaction energy arising from a potential energy that is sharply lowered, thus giving a highly-precise structure of a stable complex between the protein and the compound. There are no limitations to kinds of protein molecule system, since the program does not use a probe atom by which the ligand-binding site of the target protein is pre-treated in order to simplify energy calculations in the grid algorithm. See the preferences for Docking Study with HyperChem to the known docking program adopting the grid algorithm.
The program supports many force field calculations, i.e., MM+, Ambers, Amber2, Amber3, Amber94, Amber96, Amber99, OPLS, BIO+83, BIO+85, CHARMM19, CHARMM22, and CHARMM27 under the United Atom and All Atom conditions. Both the United Atom and the All Atom conditions can be applied to the desired parts of the protein and compound structures individually and simultaneously. These force field calculations can carry out under many minimization algorithm (Steepest Descent, Flether-Reeves, Polak- Ribiere, Newton-Raphson) supported by HyperChem, as well.
The program can assign the atomic charges of a trial compound at each conformation using the single-point calculations of a certain semi-empirical molecular orbital method. Thus, the program can perform the latest docking simulations using the atomic charges altered by the structural changes.
When the program is combined to the Gaussian Interface and ONIOM Interface of Homology Modeling for HyperChem, the program can provide the latest technology such as the QM:MM like docking simulations and the precise analysis of the obtained complex using the high-level ab initio quantum chemistry calculations such as a bond generation, a transition state analysis, and an excited state analysis.
The protein flexibility can apply to the desired parts such as some residues in the ligand-binding site, hydrogen atoms, and entire structure. In addition, the flexibility of the protein molecule system can apply to any molecules such as water molecules, small molecules, and other biological molecules. This flexibility setting is completed by selecting a part of structure using selection tool provided in the package just before simulating.
The protein system can contain any type of molecules such as metal, metal complex, small molecule, nucleic acid, and other biological molecules. The program can simulate the docking of compound, even though the ligand-binding site lies on several protein molecules.
The program can freely modify the interaction energy equation to reproduce the structure-activity relationship via the multiple linear regression analysis.
The program supports the restart function which can restart or start the docking simulations from a desired conformation without loss of the calculation precisions. The batch calculation of the docking simulations is also available when this function is used. Thus, the function is useful for searching precisely the best docking mode of the very flexible compound that has ten or more torsional bonds.
Although the program carries an algorithm optimized to the docking simulations, many high-speed techniques developed for the virtual screenings such as the loose conformation search and conformational stability test techniques are also available.
In the latest version, Docking Study with HyperChem with the multiple-compound screening function carries all functions for screening a drug candidate from a maximum 10,000 compound database.
Moreover, these advanced technologies are based on our PIEFII technology which can precisely predict the protein-ligand interaction site and the potential ligand pharmacophore and scaffold.
The following figures show the results of the PIEFII program for the cytochrome C. The second figure from the left hand side shows the predicted interaction points. The third figure from the left hand side shows the superposition between the predicted interaction points and the complexed protoporphyrin IX. As a result, the PIEFII program excellently reproduced a scaffold and chemical property of the protoporphyrin IX as well as the porphyrin-binding site. Moreover, the predicted interaction points excellently reproduced the position and conformation of two carboxyl moieties of the protoporphyrin IX. Thus, information obtained from the PIEFII program will lead to predict the native co-enzyme, even if the native co-enzyme is not known experimentally.
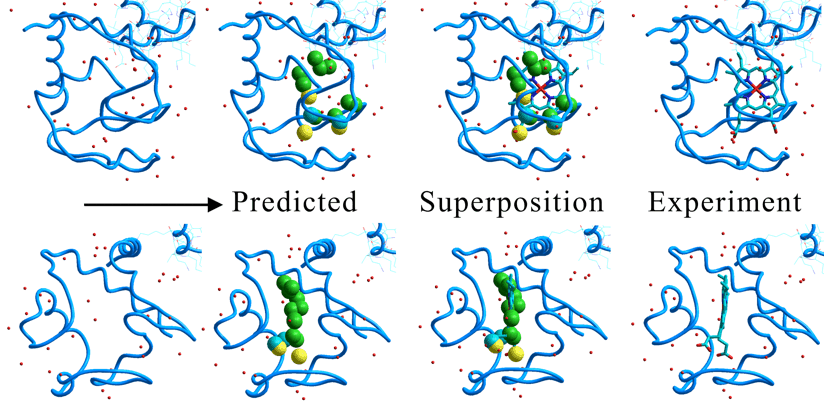
See the structure-based drug design using this program.
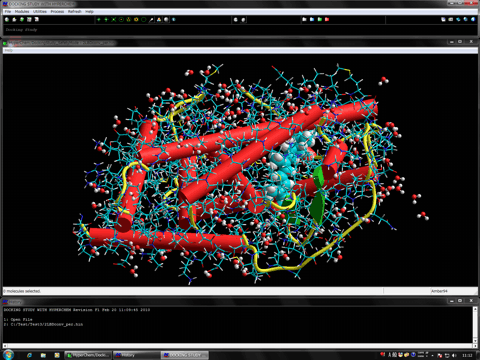
The following movie shows the result of the protein- and ligand-flexible docking simulations for a native ligand. In this study, the full conformation search for the ligand was performed under the flexible conditions of the side-chains of the protein system. In this movie, the original ligand complex to the receptor is shown in red (ball & stick) and the trial ligand obtained from the flexible docking simulations is shown in green (ball & stick). The structures of the flexible parts of the side chains are shown by tubes. In the present study, the simulations well reproduced an induced fit effect of the side chains of the receptor (In this movie, the individual receptor coordinates obtained from the docking simulations are superimposed on the original receptor coordinate).
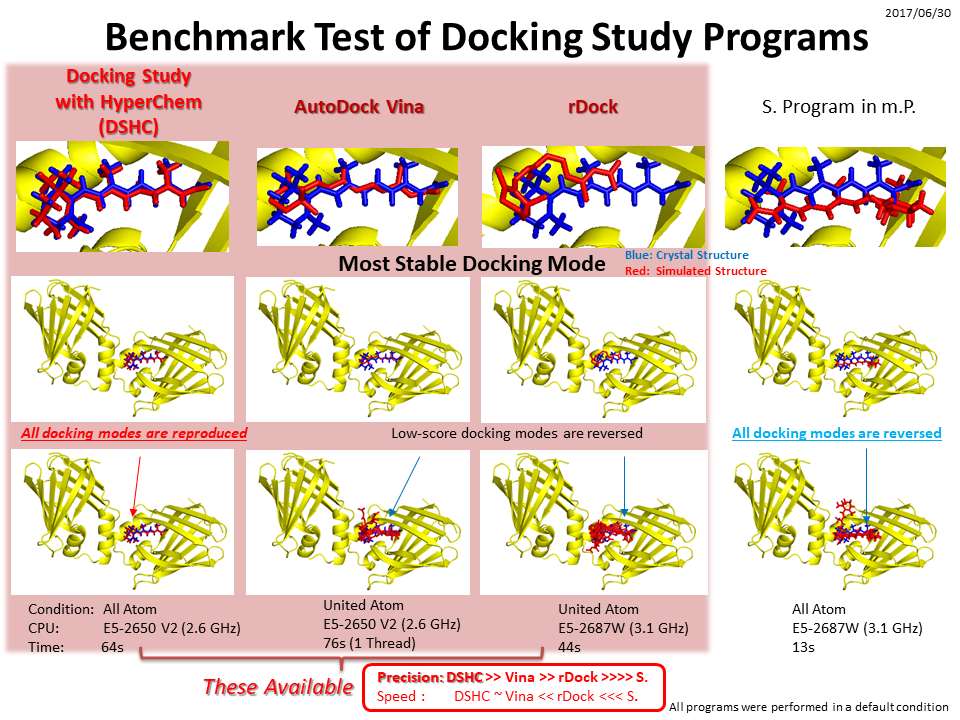
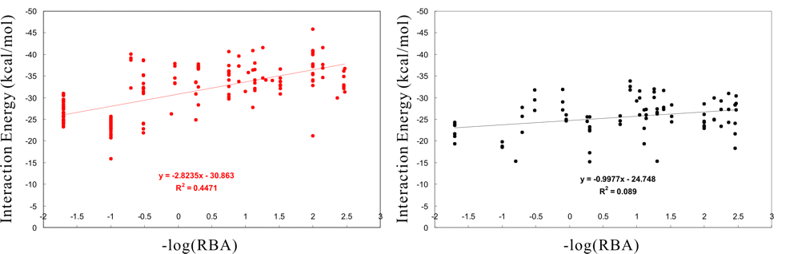
The following figure shows the comparison between Docking Study with HyperChem (left) and a known docking program based on the grid algorithm (right). These results are obtained for the same docking data set under a default setting of these programs. The figure compares their interaction energies instead of the score values due to pure comparison of these programs. As shown in figure, Docking Study with HyperChem clearly dominates to all results such as the number of hit compounds, the ratio of precisions, and the structure-activity relationships (R). See more details.
Journal of Computer-Aided Molecular Design,14, 559-572, 2000.
Most of the known docking programs including virtual screening programs are based on the grid algorithm which first calculates potential energy of a target protein molecule using some probe atoms and then accumulate the calculated energy into a lattice or center of the cells of the grid. In the grid algorithm, docking of compound performs between the compound and the grid but does not perform between the compound and the target protein molecule explicitly. However, the grid algorithm can not consider energy arising from metals and hetero molecules other than the protein molecules, since the probe atoms are optimized only for the 20 types of the amino acid residues. To date the grid algorithm was only one technique for performing docking simulations by means of the fast computations, because the docking calculation was merely sum of the energy accumulated in the individual cells of the grid. In contrast, the grid algorithm very simplifies (or approximates) the energy calculations such as long-range interaction calculations and ignores electronic and steric effects from any molecules other than a protein molecule. Therefore, the grid algorithm frequently leads to underestimation of an interaction energy and to poor results of the docking simulations. Also, a protein flexibility can not explicitly treat in the grid algorithm.
On the other hand, Docking Study with HyperChem is based on our developed PIEFII technology. Since this technology can carry out the high-speed docking simulations under the general force field calculation conditions without the grid algorithm, the program can explicitly and accurately calculate all energies arising from all atoms and molecules in the protein molecule system.
New faster conformation search algorithm is available for virtual screenings.
Docking Study with HyperChem supports docking simulation with a hypervalent compound.
The following figure shows the results of the re-docking study with a hypervalent compound. Crystal structure shows blue tube and Simulated structure shows red tube.
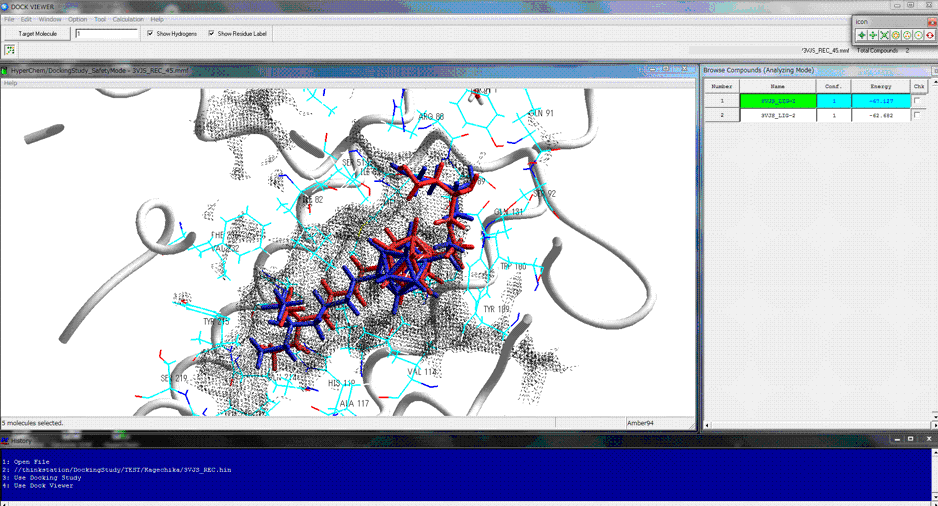
Supported the novel flexible docking algorithm which can be used for docking compound to a site in a large structural change exceeding the induced fit effect of the biological molecule such as the structural changes between the apo and holo conformations.
The following figure shows the results of the protein- and ligand-flexible docking simulations using the novel docking algorithm. In the apo conformation, the potential ligand-binding site is destroyed or occupied by its inner structure. The comprehensive and automatic docking simulations of the native ligand for the target adopting the apo conformation well reproduced the structure of the experimentally solved stable complex. In this figure, red shows the stable complex obtained from the docking simulations and blue shows the experimentally solved complex in the corresponding holo conformation.
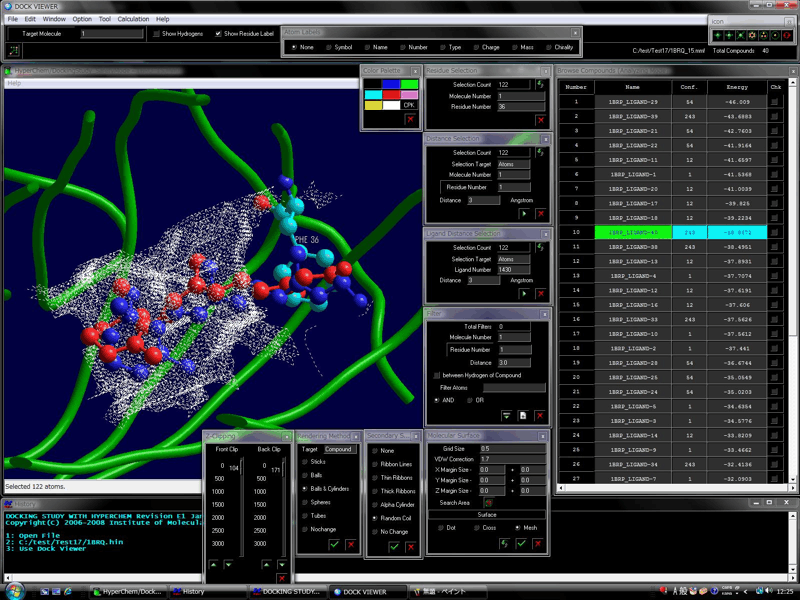
The automatic biological macromolecule and peptide docking simulation is also available using the Automatic Conformation Search method.
The following figure shows the results of the docking simulations between the antibody and peptide. The peptide recognition region positions between the heavy chain and light chain. Red structure (ball & stick) shows the most stable complex obtained from the docking simulation and blue structure (ball & stick) shows the experimentally solved complex. In the right-hand figure, the peptide recognition region (ball & stick) of the antibody was treated as a flexible part.
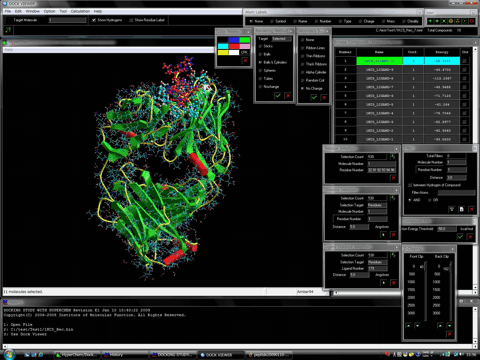
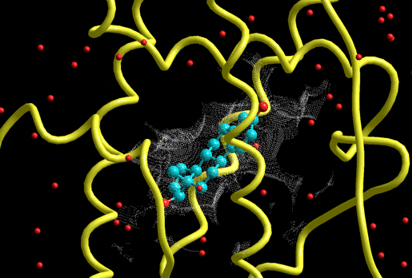
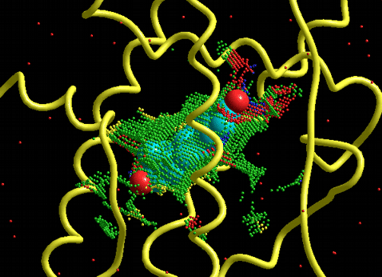
Supported the automatic nucleic acid- and ligand-flexible docking simulations as well as the protein- and ligand-flexible docking simulations.
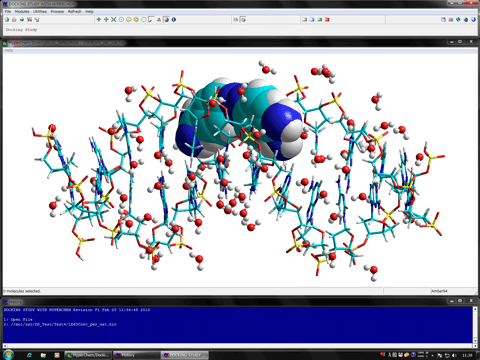
The following figure shows the results of the nucleic acid- and ligand-flexible docking simulations using the DNA and the minor groove binder. Green structure shows the most stable complex obtained from the docking simulations and purple structure shows the experimentally solved complex.
Other Feature
The program carries on the PIEFII technology in the next-generation drug discovery.
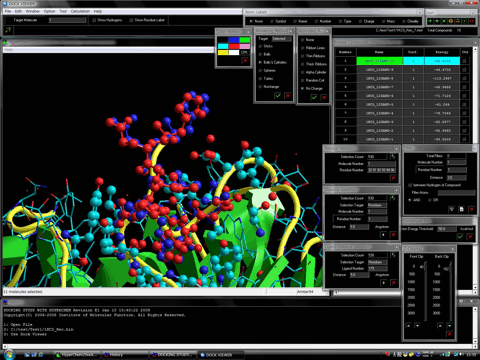
The package contains a specialized multifunctional browser, Dock Viewer, for viewing the result of the docking simulations.
An initial structure of a trial compound can be automatically prepared using Mol Dimension program.
Mol Browser can be used for browsing a 3D structure of a trial compound.
The Docking Study module program can automatically assign information on a trial compound such as a torsion and ring system. The torsion editor interface is useful for preparing the individual rotation angles.
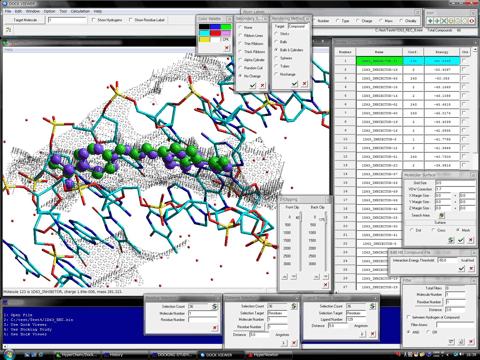
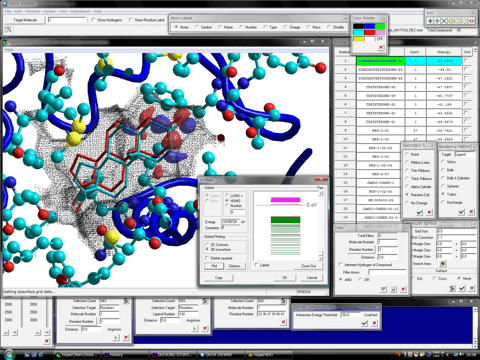
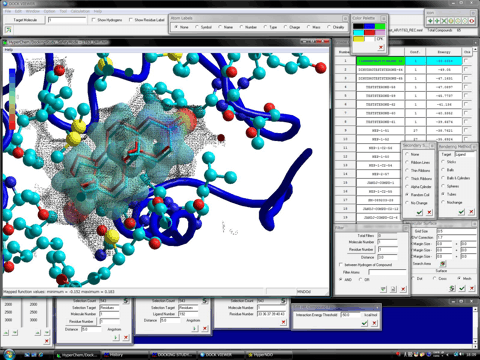
The structure of the stable complex between the trial compound and the protein system is represented by the OpenGL function of HyperChem (version 7.5 or the later). The browsing of the stable complexes is carried out under the fully-automatic rendering function in the level of a demonstration or a figure of a literature. In addition, the program can display the molecular surface (van der Waals surface) at a certain region such as the ligand-binding site together with the hit compounds. The following figures show the mesh surface (left) and the dot surface with the corresponding atom color (right), respectively.
Dock Viewer program can simultaneously represent any molecular properties, e.g., molecular orbitals, charge densities, and electrostatic potentials, of a hit compound, rendering in many style. For example, the following figures show a hit compound formed the most stable complex with a target protein. At left-hand side of the figures, the highest occupied molecular orbital (HOMO) of the hit compound is shown together with a native ligand (white tubes) and the molecular surface (black mesh). At right-hand side of the figures, the isosurface of the electrostatic potentials of the hit compound is shown. Additionally, ONIOM Interface for Receptor in the Homology Modeling Professional for HyperChem package can analyze interactions of the stable complex between a hit compound and a target protein molecule using the high level ab-initio quantum chemistry calculations.
Pamphlet of Docking Study with HyperChem, Revision H1 (Japanese, PDF: 1 MB)
Pamphlet of Docking Study with HyperChem, Revision G1 (Japanese, PDF: 2 MB)
Pamphlet (Japanese; 5 MB) for earlier version
SBDD Pamphlet (PDF: 2 MB; Japanese)
References
Motonori Tsuji, Molecular Science. 1, NP004, 2007.
Science. 319, 624-7, 2008.
Biochemical and Biophysical Research Communications. 339, 173-177, 2010.
Biochemistry. 49, 10647-10655, 2010.
Plant Cell Physiol. 53, 1638-1647, 2012.
Motonori Tsuji, Journal of Structural Biology. 185, 355-365, 2014.
Motonori Tsuji, et. al., Journal of Computer-Aided Molecular Design. 29, 975-988, 2015.
Motonori Tsuji, Journal of Molecular Graphics and Modelling.62, 262-275, 2015.
Motonori Tsuji, et. al., FEBS Open Bio 7, 391-396, 2017. DOI: 10.1002/2211-5463.12188.
Motonori Tsuji. Journal of Computer-Aided Molecular Design. 31, 577-585, 2017. DOI: 10.1007/s10822-017-0025-6.
J. Pharmacol. Exp. Ther. 372, 277-284, 2020.
Motonori Tsuji, FEBS Open Bio 10, 995-1004, 2020.
J. Virol. 94, e00252-20, 2020.
Chem. Pharm. Bull. 68, 1193-1200, 2020.
PLOS ONE 16, e0257705, 2021.
Motonori Tsuji. Int. J. Mol. Sci. 23, 11009, 2022.
The most powerful and sophisticated molecular modeling environment
HyperChem front-ended
Module Programs
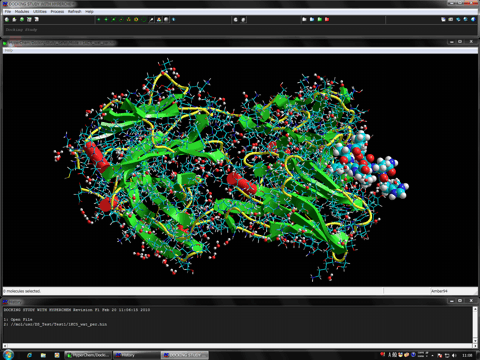
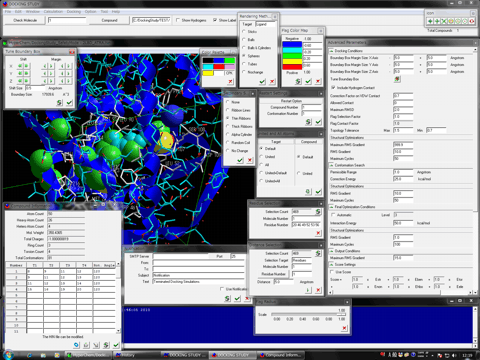
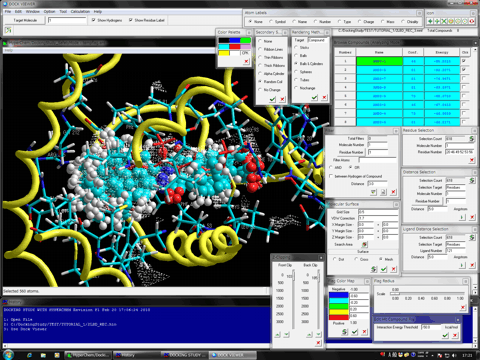
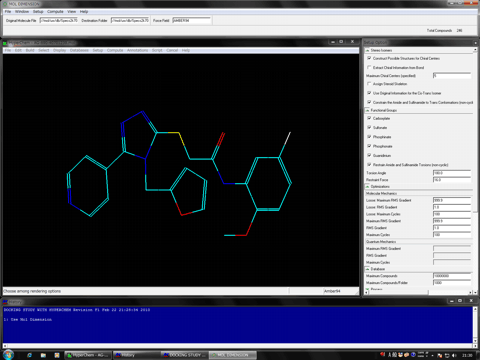
AutoDock Vina In Silico Screenings Interface
| Family | PIEFII | Docking Study | Dock Viewer | Mol Dimension | Mol Browser | AutoDock Vina Screening*** |
| Essential | Limited* | Single Compounds | None-Limited | Single Compounds | None-Limited** | None-Limited |
| Premium Essential | Limited* | Max. 10 Compounds | None-Limited | Max. 10 Compounds | None-Limited** | None-Limited |
| Professional | Limited* | Max. 100 Compounds | None-Limited | Max. 100 Compounds | None-Limited** | None-Limited |
| Advanced | Limited* | Max. 1,000 Compounds | None-Limited | Max. 1,000 Compounds | None-Limited** | None-Limited |
| Ultimate | Limited* | Max. 10,000 Compounds | None-Limited | Max. 10,000 Compounds | None-Limited** | None-Limited |
| Cluter Virtual Screening System |
None-Limited | None-Limited, Cluster | None-Limited | None-Limited, Cluster | None-Limited | None-Limited |
* Simultaneous predictable maximum atoms: 1000 atoms, ca. 80 residues.
** Although the program is non-limited version, the available number of compounds depends on the Docking Study and Mol Dimension module programs.
*** No limitation of number of compounds for AutoDock Vina screenings.
Add-on Options for Docking Study with HyperChem*
| Docking Study module: Conventional Screening Option (The number of compounds is not limited)
Mol Dimension module: Conventional Screening Option (The number of compounds is not limited) |
* The Conventional Screening Option is unable to use the compound conformation search function and the flexible option of receptor. In the Conventional Screening Option, the individual conformations of compound must be prepared as a separated trial compound. The Conventional Screening Option does not support the Clustering function. The Clustering function requires Virtual Screening System.
Recommended Minimum System Requirements
Processor: Multi-Core Intel Core or Xeon 2 GHz (Intel Pentium III or later)
Operating System: Microsoft Windows 10 and 11 (Microsoft Windows 98 or later, XP is necessary for all function)
Memory (RAM): 8 GB (Minimum 256 MB)
Monitor: SXGA or more
Video Card: OpenGL board recommended
Mouse: Multi-functional mouse
Other: HD 512 GB or more (2TB or more for treating big (100 million compounds) SDF and/or MOL2 files); CD-ROM drive; Network card
Software Requirements
Essential
HyperChem (Windows version; except for Student version): HyperChem Release 8.0.10 Professional for Windows (Release 5.1.1 or later, Release 7.5.2 is necessary for all function)
TclPro1.2 (Windows version): TclPro1.2 for Windows is necessary to run Homology Modeling Professional for HyperChem. TclPro1.2 is available on the Web free of charge.
Option
AutoDock Vina (Windows version): AutoDock Vina program is necessary to perform AutoDock Vina in silico screenings.*
* Note that AutoDock Vina program is different docking program from Docking Study with HyperChem. Docking Study with HyperChem only provides the useful GUI environment for performing the AutoDock Vina in silico screenings. No limitation of number of compounds for AutoDock Vina screenings.
OpenBabel (64bit or 32bit Windows version): OpenBabel for Windows is necessary for exporting in MDL SDF, Tripos MOL2, or AutoDock PDBQT format from HIN (HyperChem format) or PDB format.
Other
NAMD and VMD is necessary for performing ns scale molecular dynamics. ABINIT-MP and BioStation Viewer or GAMESS and Fu orFacio can be used for performing quantum mechanics calculations for entire molecular system.
Remote Desktop and Multi-Core Environment
Docking Study with HyperChem can simultaneously run on the number of CPU cores, so that one PC workstation with some CPU cores can perform the clustering function.
The Cluster version of Docking Study with HyperChem prepares the automatic clustering function among the multiple PC workstations using Remote Desktop function of Windows.
Installing the Cluster version into multiple PC workstations with HyperChem Site License version, you can perform the virtual screenings for compound database consisting of millions or more number of compounds.
Description
Compatible to the Protein Data Bank format versions 3.0-3.3.
Docking Study with HyperChem is a package of Tcl/Tk programs (mainly user interface) and 32bit Windows console applications.
These programs are compiled by the TclPro1.2 compiler. Thus, in order to run the package, you must install TclPro1.2 in your Windows system, prior to the installation of the package.
A substantial Online-Help (English) is available from individual programs in the package.
The package includes two user's manuals (presently Japanese Language Only).
Manual Contents (Japanese Language)
Manual 2 Contents (Japanese Language)
Important: When you installing the software, please make sure that HyperChem and TclPro1.2 have been installed in your system.
* HyperChem is a registered trademark of Hypercube, Inc.