

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
Molecular Modeling and Structure-based Drug Design Systems
ONIOM Interface for Receptor
- Full Automatic Multi-Layer ONIOM Interface -
Compatible to Gaussian98, Gaussian03, Gaussian09, Gaussian16 & GaussView3, GaussView4, GaussView5, GaussView6
In quantum mechanics calculations for the entire structure of big molecular system such as biomacromolecular system, the single point calculations for the initial structure obtained from classical molecular mechanics calculations and/or classical molecular dynamics calculations cannot converge or will give abnormal energies. For obtaining reliable results from quantum mechanics calculations such as fragment molecular orbital methods of entire molecular system, the initial structure must be prepared using geometrical optimization calculations via ONIOM methods. ONIOM Interface for Receptor can provide the best solution for the quantum mechanics calculations of the entire molecular system of which the precise initial structure is prepared from Docking Study with HyperChem and Homology Modeling Professional for HyperChem.
Motonori Tsuji, et. al. FEBS Open Bio., 7, 391-396, 2017, DOI: 10.1002/2211-5463.12188.
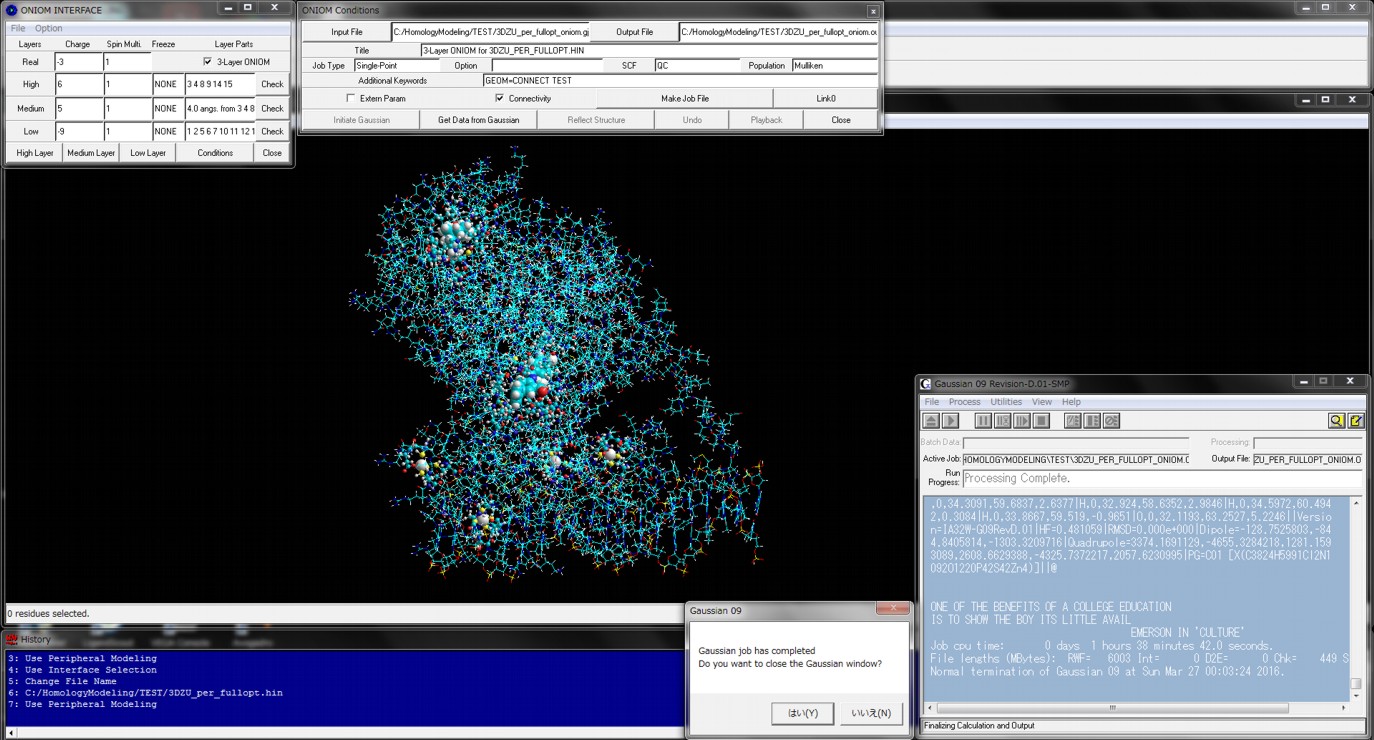
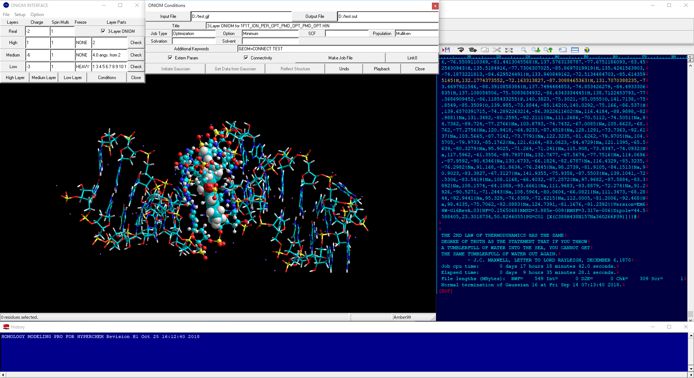
Figure shows the result of ONIOM single-point calculations (RHF/6-31G:RAM1:AMBER) for the whole system which is constructed of a hetero dimer of two nuclear receptors (including ligand-binding domain and DNA-binding domain), hormone response elements of DNA, and cofactor fragments. In this calculation, two types of small compounds and four zinc atoms are set to the high layer (depicted in spheres), the protein residues, nucleotides, and water molecules around 4.0 angstrom from the high layer are set to the medium layer (depicted in balls and cylinders), and the other structures are set to the low layer (depicted in sticks). Since ONIOM Interface can automatically prepare all parameters, Gaussian program can be initiated simultaneously after starting the ONIOM Interface.
The most powerful and sophisticated molecular modeling environment
HyperChem front-ended
The most powerful computational chemistry environment
Gaussian back-ended
Feature & Function
At present, the ONIOM method of Gaussian, the MOZYME method of Mopac200X, and the ab-initio fragment MO method are the representative quantum chemistry calculations for calculating a big molecular system such as a protein molecule system. ONIOM Interface for Receptor is an interface for performing the multi-layer ONIOM calculations of Gaussian using the HyperChem. On the other hand, an interface, Mopac Interface for HyperChem, to the MOZYME method is available for HyperChem. Furthermore, the latest version is compatible to the fragment molecular orbital calculation programs such as ABINIT-MP (BioStation Viewer) andGAMESS (Fu / Facio) programs. Thus, all three representative quantum chemistry calculation methods become available for calculating the big molecular system using the HyperChem graphical user interface.
ONIOM Interface for Receptor can automatically create a Gaussian job file for performing the 2-layer (QM:QM and QM:MM) and 3-layer (QM:QM:QM and QM:QM:MM) ONIOM calculations of Gaussian using the excellent HyperChem molecular modeling functions. The results (atomic charge and the structure) of the Gaussian calculations can be reflected to the molecular system in the HyperChem workspace immediately.* The interface can deal with a molecule system, which consists of biological macromolecules (proteins and/or nucleic acids) and small molecules such as a receptor and a ligand, and can automatically create the missing parameters of force field, the link atoms, and the charges.** Thus, as soon as the program is started, the Gaussian job can be initiated after several seconds without any manual parameter settings, regardless of the preparation of the job file for the 2-layer or 3-layer ONIOM calculation for the big molecular system (the program is also very useful for preparing the Gaussian job file of the UNIX 64 bit platforms.).
ONIOM Interface for Receptor and Gaussian Interface for HyperChem are the add-on programs to the Peripheral Modeling Professional module program. Thus, this program can operate a molecular system under the quantum chemistry conditions.
See examples.
* When a checkpoint file name is specified in the Link0 entry box of ONIOM Interface for Receptor, the results such as the molecular orbitals and electrostatic potentials of the corresponding Gaussian job can be analyzed in detail, using a Gaussian viewer through the checkpoint or formcheck file prepared.
** In cases of (QM:MM) and (QM:QM:MM), only Amber force field is available for the molecular mechanics method in the low layer model chemistry.
The following figures show the results of superposition between the initial structure (blue) and the optimized structure (red). The Gaussian job was performed at the 2-layer ONIOM(HF/6-31G*:Amber) model chemistry.

The following figures show the results of superposition between the initial structure (red) and the optimized structure (blue). The initial structure was prepared using the Peripheral Modeling Professional program and the optimized structure was extracted from the Gaussian job file using the add-on ONIOM Interface for Receptor. The Gaussian job was performed at the 2-layer ONIOM(HF/6-31G*:Amber) model chemistry. Figure at the right hand side shows the superposition between the initial structure and the optimized structure obtained from the partial optimizations using frozen atom conditions for the heavy atoms in the low layer parts of the receptor.


In the latest version, ONIOM calculations of a complex molecular system can be performed by one click. Of course, ONIOM calculations for a complex structure registered into PDB database as well as the stable complex structure obtained from the docking simulations of Docking Study with HyperChem can be performed. The ONIOM optimized structures can be used for quantum chemistry calculations using fragment molecular orbital calculations.
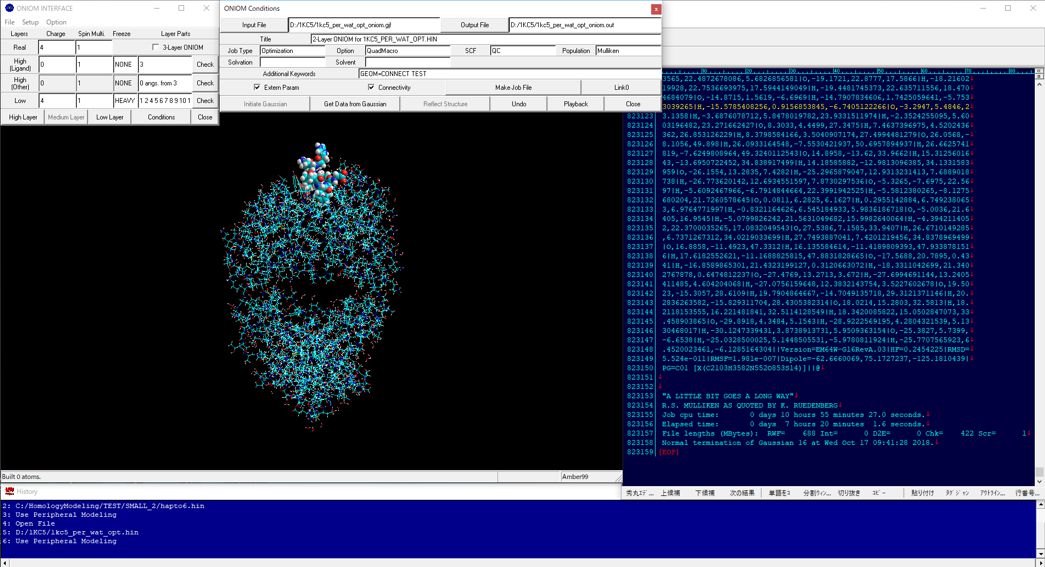
The following figure shows the results of 2-layer ONIOM calculations for antigen-antibody system. In this case, an antigen was set to the high-layer (HF/6-31G) and fully optimized, and 2 antibody and 176 crystal water molecules were set to the low-layer (AMBER) and partially optimized for hydrogen atoms.
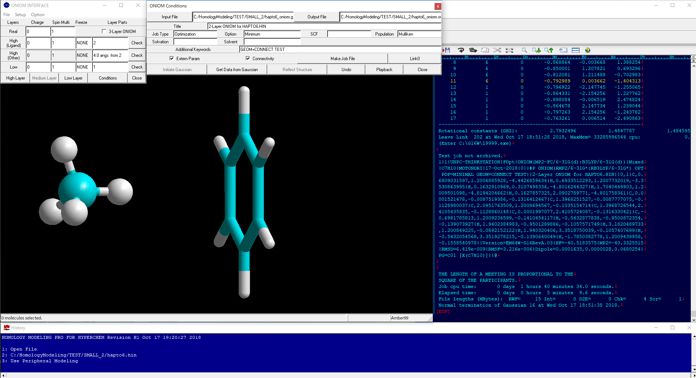
The following figure shows the results of the 2-layer ONIOM calculations for the hapto-6 type interaction between methane and benzene molecules. In this case, methane was set to the high-layer (MP2/6-31G*) and benzene was set to the low-layer (B3LYP/6-31G*), and fully optimized.
The following figure shows the 3-layer ONIOM calculations for the DNA-medium size molecule system. In this case, the medium-size molecule was set to the high-layer (HF/6-31*), and fully optimized. The nucleotides around 4 angstrom from the medium-size molecule were set to the medium-layer (AM1) and other parts were set to the low-layer (AMBER), and partially optimized for hydrogen atoms.
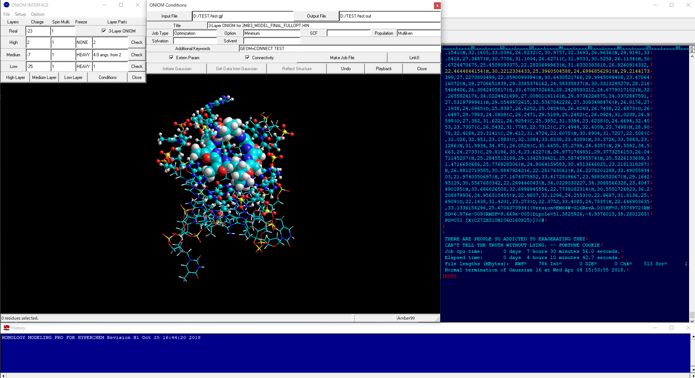
The following figure shows the 3-layer ONIOM calculations for the RNA-small molecule system. In this case, the small molecule was set to the high-layer (B3LYP/6-31*), and fully optimized. The nucleotides around 4 angstrom from the small molecule were set to the medium-layer (AM1) and other parts as well as counter ions were set to the low-layer (AMBER), and partially optimized for hydrogen atoms.
References
Motonori Tsuji, Molecular Science. 1, NP004, 2007.
Science. 319, 624-7, 2008.
Biochemical and Biophysical Research Communications. 339, 173-177, 2010.
Biochemistry. 49, 10647-10655, 2010.
Plant Cell Physiol. 53, 1638-1647, 2012.
Motonori Tsuji, Journal of Structural Biology. 185, 355-365, 2014.
Motonori Tsuji, et. al., Journal of Computer-Aided Molecular Design., 29, 975-988, 2015.
Motonori Tsuji, Journal of Molecular Graphics and Modelling., 62, 262-275, 2015.
Motonori Tsuji, et. al., FEBS Open Bio., 7, 391-396, 2017. DOI: 10.1002/2211-5463.12188.
Motonori Tsuji. Journal of Computer-Aided Molecular Design., 31, 577-585, 2017. DOI: 10.1007/s10822-017-0025-6.
J. Pharmacol. Exp. Ther. 372, 277-284, 2020.
Motonori Tsuji. FEBS Open Bio., 10, 995-1004, 2020.
J. Virol. 2020.
Chem. Pharm. Bull. 68, 1193-1200, 2020.
PLOS ONE 16, e0257705, 2021.
Motonori Tsuji. Int. J. Mol. Sci. 23, 11009, 2022.
Description
ONIOM Interface for Receptor has been integrated into the Peripheral Modeling Professional module program in the Homology Modeling Professional for HyperChem package. Refer also to the Peripheral Modeling Professional module program.
ONIOM Interface for Receptor is available for the molecular system which consists of at least two or more molecules.
New, High-Performance Software for Biomacromolecule Modeling, Functional Analysis, and Simulation.
Homology Modeling Professional for HyperChem
with Gaussian Interface for HyperChem & ONIOM Interface for Receptor
Compatible to NAMD Molecular Dynamics and ABINIT-MP / GAMESS Fragment Molecular Orbital Quantum Mechanics Calculations for Entire Molecular System
* HyperChem is a registered trademark of Hypercube, Inc.
** Gaussian is a registered trademark of Gaussian, Inc.