

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
Docking Study
Docking Algorithm
A compound is superposed on the ligand-binding site of a target protein in terms of the structure-based manner.
The superposition is carried out using flags or grids representing a certain chemical property of the compound and the ligand-binding site (this chemical property brings about individual properties of docking programs).
In general, the ligand-binding site of a target protein is treated as a static state. In this case, an induced fit effect and any effects of protein side chains are approximated by the force field potentials.
Recently, a flexible docking which simultaneously considers an induced fit effect of the side chain in a ligand-binding site is becoming mainstream in the docking study. Note that most of the docking programs are termed as the FLEXIBLE docking program, although they treat only the compound flexibility.
Once superposition succeeds, each torsional bond of the superposed compound in an initial structure is rotated at a certain rotation angle (30°, 45°, 60°, 90°, or 120°). Subsequently, the interaction energy for all docking modes accompanying to the rotations of the torsional bonds is calculated using the structural optimizations. In this docking algorithm, the number of simulations is reduced in but the accuracy of dockings is lowered. To avoid this problem, the docking program carrying this algorithm needs to prepare some initial structures per single compound.
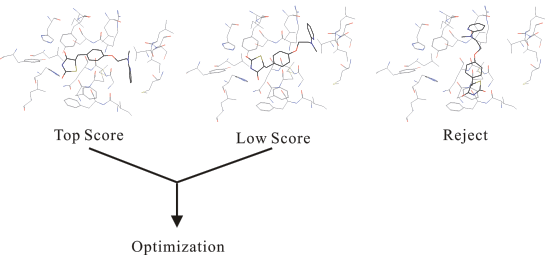
After being optimized, the individual docking modes are assessed by an energy function which is carried in the individual docking programs and has been tuned for the current target protein. In general, most of the docking program adopting the grid algorithm calculate a score value rather than the interaction energy, since the grid algorithm largely simplifies the energy calculations.
A compound that adopts higher score in these modes is suggested as a candidate for the target protein molecule.
Grid Algorithm
Most of the known docking programs including virtual screening programs are based on the grid algorithm which first calculates potential energy of a target protein molecule using some probe atoms and then accumulate the calculated energy into a lattice or center of the cells of the grid. In the grid algorithm, docking of compound performs between the compound and the grid but does not perform between the compound and the target protein molecule explicitly. However, the grid algorithm can not consider energy arising from metals and hetero molecules other than the protein molecules, since the probe atoms are optimized only for the 20 types of the amino acid residues. To date the grid algorithm was only one technique for performing docking simulations by means of the fast computations, because the docking calculation was merely sum of the energy accumulated in the individual cells of the grid. In contrast, the grid algorithm very simplifies (or approximates) the energy calculations such as long-range interaction calculations and ignores electronic and steric effects from any molecules other than a protein molecule. Therefore, the grid algorithm frequently leads to underestimation of an interaction energy and to poor results of the docking simulations. Also, a protein flexibility can not explicitly treat in the grid algorithm.
May 2005
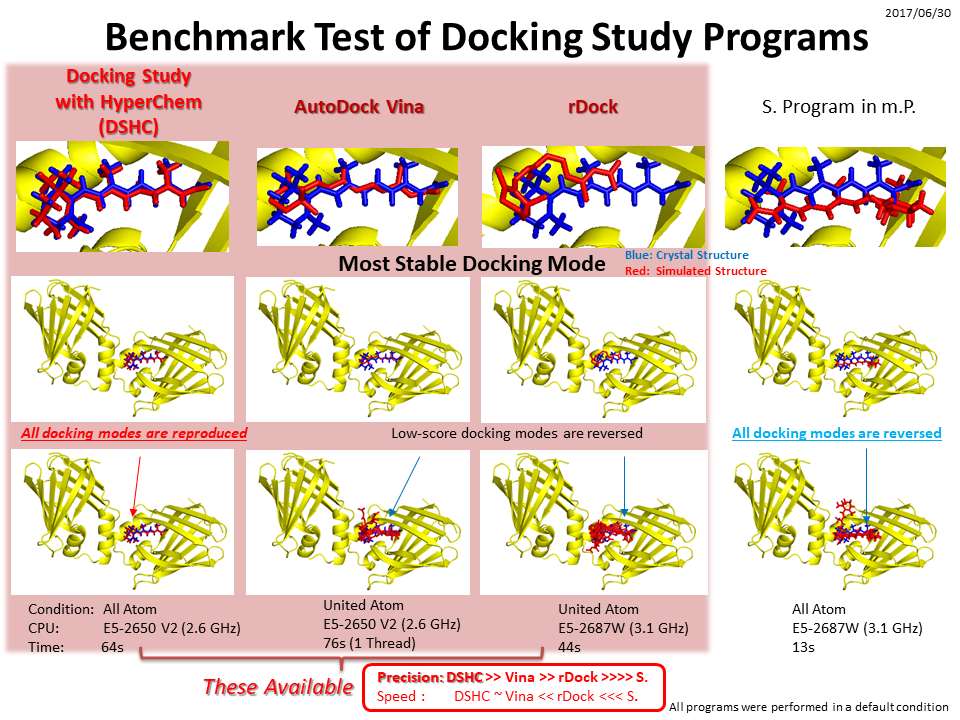
See our biological macromolecule- and ligand-flexible docking and screening program, Docking Study with HyperChem, which carries out a novel and most powerful docking algorithm.
Return to the Virtual Screening