![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
Homology Modeling Professional for HyperChem
Click on the individual program names to access to their detailed features.
![]()
Homology Modeling Professional
Feature
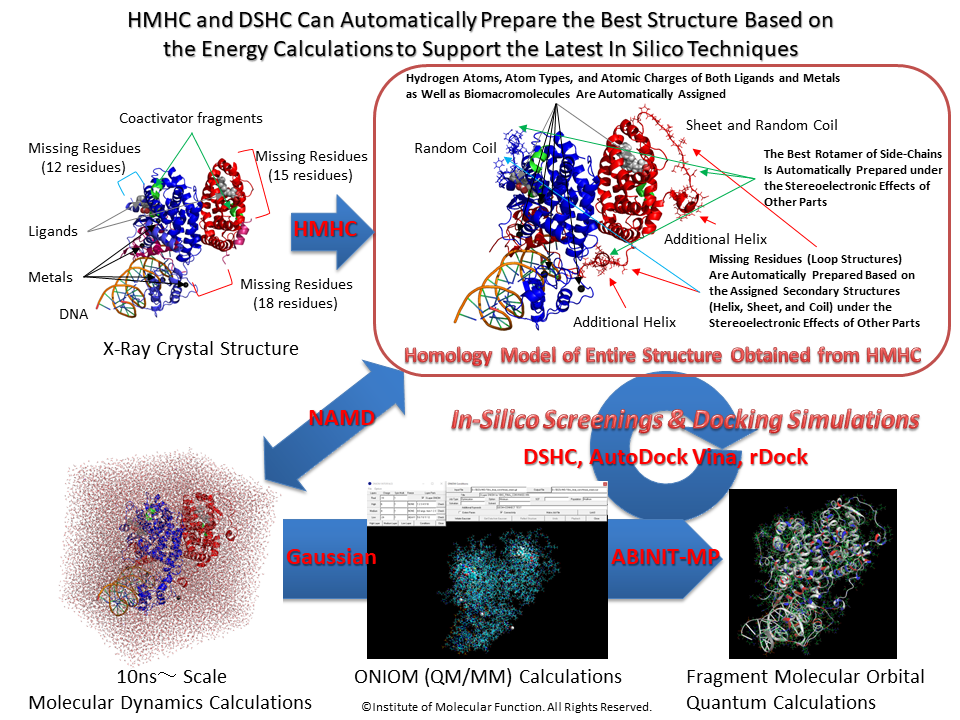
The "Homology Modeling" program automatically creates a three-dimensional (3D) structure of a target sequence using a template (or reference) 3D structure in the HyperChem workspace (See homology modeling approach). The program carries a novel logical method for aligning sequences. That is, a secondary structure information obtained from a certain prediction program can be used for optimizing a sequence alignment (This module program can carry out the highly-precise secondary structure prediction and the ab init 3D structure prediction programs, which were developed by us). For aligning sequences, the program supports many score matrices. In addition, a protein model can be constructed keeping a protonation state assigned for the individual histidine residues and a conserved S-S bond.
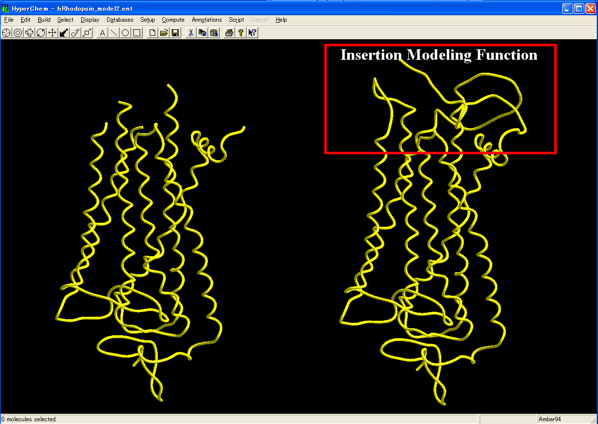
Moreover, the program carries a novel Insertion Sequence Modeling function which can easily create a structure for a certain insertion sequence with infinite length. This function can also coordinate a secondary structure such as a helix and a sheet into the created structure as well as can construct a secondary structure continuing to a known secondary structure. A structure of an insertion sequence can be created on the basis of the above secondary structure predictions, as well. The created structure of an insertion sequence can be optimized through several steps using the molecular mechanics calculations under a desired force field condition, i.e., Ambers, Amber2, Amber3, Amber94, Amber96, Amber99, MM+, OPLS, Bio+83, Bio+85, CHARMM19, CHARMM22, CHARMM27.
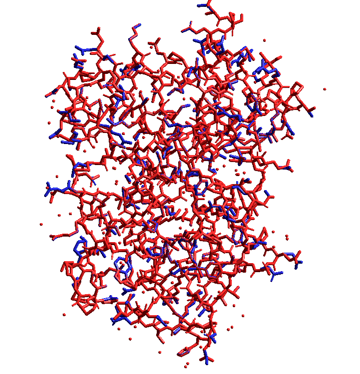
A best conformation (i.e., rotamer) of a residue side chain can be logically and automatically determined using the ultimate rotamer modeling function of the Side-Chain Rotamer Modeling module program in the package.
The following figure shows the results for the auto-rotamer search function of the Side-Chain Rotamer Modeling module program. In this figure, the experimental crystal structure is depicted in blue and the predicted structure is depicted in red. Although some rotamers of the charged residues on the protein surface differed each other due to equilibrium among some conformations, other rotamers excellently reproduced the experiment.
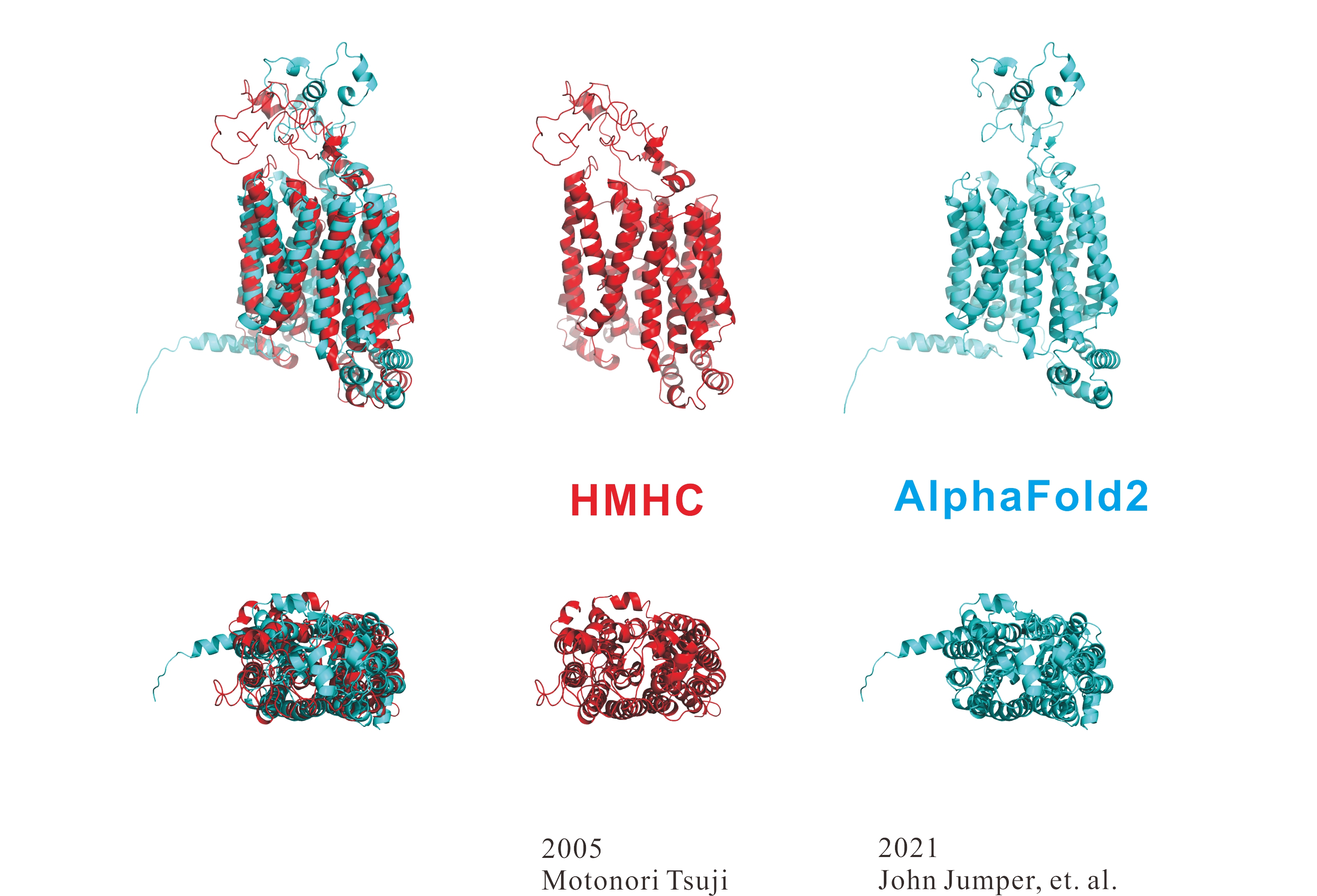
AlphaFold2 vs Homology Modeling Professional for HyperChem (HMHC)
The following figure shows the results of three-dimensional structure prediction for an unknown protein sequence for 20 years including family proteins, using HMHC and AlphaFold2.
HMHC can provide a precise three-dimensional structure since HMHC can model the side-chain rotamer conformations and the interactions between other bioamacromolecules and small molecules as well as crystal waters.