

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
Homology Modeling Professional for HyperChem
Click on the individual program names to access to their detailed features.
![]()
Side Chain Rotamer Modeling Professional
Feature
The "Side Chain Rotamer Modeling" program can provide all of the rotamer-modeling functions, the manual modeling, the database modeling, the auto-rotamer search modeling, and their combined modelings. The auto-energy estimation function (using the partial geometry optimizations and single-point calculations) is available for the side chain rotamer modeling. This function supports the Ambers, Amber2, Amber3, Amber94, Amber96, Amber99, OPLS, MM+, Bio+83, Bio+85, CHARMM19, CHARMM22, and CHARMM27 force field conditions, depending on your HyperChem version (see Calculation Results). Both faster method and conventional method are available for the search. The faster method supports both the partial geometry optimization calculations and the single-point calculations, and the conventional method supports the single-point calculations. Refer to the Calculation Times for a practical computational time. Alternatively, the program can treat a side chain rotamer database. The latest database is available for the side chain modeling.* Since the auto-energy estimation function (geometry optimizations and single-point calculations) can be used for the database modeling of the rotamers, a combination of these functions will provide the best method for performing the rotamer modeling of the big molecular system (tens of thousands of atoms).** See also the results of the auto-rotamer search modeling using the latest rotamer database.
Moreover, the batch calculations are supported for the auto-rotamer search modeling (database mode and non-database mode). The batch calculations can search the most stable rotamer of all residues, which are not identical to the corresponding residues of the template protein, toward the C-terminal residue from the N-terminal residue in default. The order of the batch calculations can freely exchange each other. Thus, the batch calculations can be applied to a model which shows very low amino-acid identity with the template and whose side chain is surrounded by many non-estimated side chains. The batch calculation is also available for some selected amino-acid side-chains as well as all side chains.
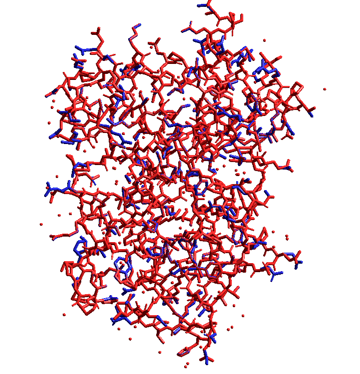
The following figure shows the results (about one hour) for the auto-rotamer search function of the Side-Chain Rotamer Modeling module program (using the faster method of the single point calculations and the database mode under the Amber94 force field). In this figure, the experimental crystal structure is depicted in blue and the predicted structure is depicted in red. Although some rotamers of the charged residues on the protein surface differed each other due to equilibrium among some conformations, other rotamers excellently reproduced the experiment.
* The new rotamer database is based on the statistical analysis of about 4,000 X-ray crystal structures (without overlap of the same proteins in about 25,000 structures) in the range of 0.0-4.0Å resolutions.
** This method maximally saves 95% computation time as compared with the auto-rotamer search with rotation angles (60°) in default.