![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
検証報告
Side Chain Rotamer Modelingプログラム
検証
人プロゲステロンレセプター(PR) リガンド結合領域 (LBD) (1A28; Molecule B)を用いてホモロジーモデリングされた人アンドロゲンレセプター(AR)LBDの側鎖ロータマーモデリング
結果
今回の検証は一点計算を用いたエネルギー評価であったが、"Side Chain Rotamer Modeling"プログラムはhAR LBDの側鎖ロータマーを良好に再現した(ただし、hAR LBDとhPR LBDの間の異なった主鎖コンフォメーションに起因するGln783のロータマーを除く)(下図)。したがって、標準装備されている構造最適化を用いたエネルギー評価では更なる予測の向上が期待される。
さらに、ロータマーデータベースを使用した自動ロータマー探索機能を用いた場合には、同条件にて92%作業時間短縮で全探索を完了した(非描画モードではさらに計算時間が短縮される)。また、本テストはHyperChem6.03で実施したが、HyperChem7.51ではさらに半分の計算時間で全探索が完了した。
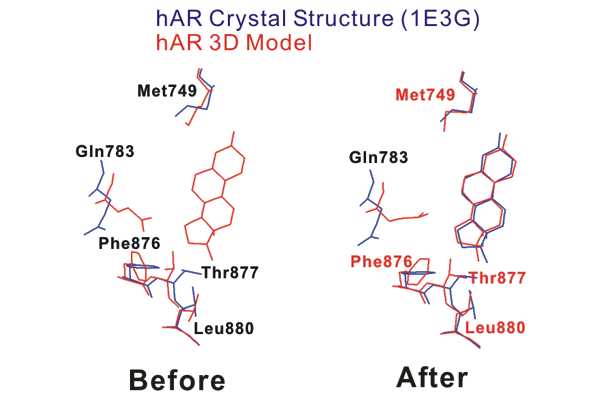
図. hAR LBDの結晶構造(青)とモデル(赤)の重ね合わせ
リガンド結合部位(リガンドから5Å以内)にあるhAR LBDとhPR LBD(鋳型)の間で異なる残基(5残基)がこの図に示されている。hAR LBDの3Dモデルは赤で、hAR LBDの結晶構造が青で示されている。hAR LBDの初期3Dモデル(手順3)と結晶構造の重ねあわせが左手("Before")に示されている。hAR LBDのロータマーモデリングされた3D構造と結晶構造の重ねあわせが右手("After")に示されている。
条件
鋳型との同一性: 54.8 %
鋳型と異なる残基数: 112
アミノ酸長: 249
リガンド: メトリボロン (R1881)
手順
1. Homology Modeling Program
モデル: hPR LBDの鋳型3D構造を用いたhAR LBD 3Dモデル作成
2. HyperChem
N- および C-末端: ツビッターイオン (INDO電荷)
リガンド: MNDO/d電荷, Amber94原子タイプ
3. HyperChemおよびInterface Selection Program
力場: Amber94
計算: 全水素原子に対する構造最適化 (勾配エラー、0.1)
4. Side Chain Rotamer Modeling Professional (バッチ計算)
力場: Amber94
Faster法, 再描画: ON, その他: デフォルト
自動ロータマー探索機能の場合
二面角: gamma = 60°, delta = 60°, epsilon = 60°, zeta = 60°
全ロータマー: 24,059 (24,059一点計算)
データベース自動探索機能の場合
全ロータマー:2,111(2,111一点計算)
5. HyperChem
計算: 全側鎖に対する構造最適化 (勾配エラー、1.0)
6. Restraints Program
最終座標: 上記作成 hAR 3Dモデル
束縛: レセプターの主鎖と側鎖重原子("All"オプション)およびリガンド重原子
束縛力: デフォルト
7. HyperChem
束縛条件下の低温(300 ケルビン, 3 ピコ秒)分子動力学アニーリング
使用システム環境
CPU: インテル Pentium4 3.06G (FSB 533MHz; L2 512KB)
メモリー: PC1066 1GB ECC
チップセット: インテル I850E
SCSI: Ultra320
HDD: Ultra320 17GB
OS: マイクロソフト Windows XP Professional SP2
HyperChem: バージョン6.03
Homology Modeling Professional for HyperChem, Rev. A2
2005年6月6日