主要研究概要
Motonori Tsuji*. Potential Anti-SARS-CoV-2 Drug Candidates Identified through Virtual Screening of the ChEMBL Database for Compounds That Target the Main Coronavirus Protease. FEBS Open Bio, 10, 995-1004, 2020.
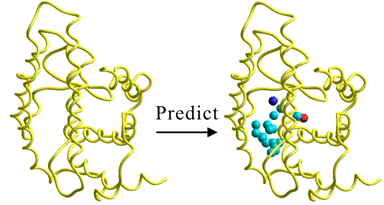
新型コロナウイルス(重症急性呼吸器症候群コロナウイルス2型[SARS-CoV-2])はCOVID-19の病原ウイルスとして同定された。SARS-CoV-2はその強力な飛沫、接触、あるいは空気感染力により世界中に蔓延し、パンデミックを引き起こした。本概要執筆時(2021年5月4日)で世界の感染者数は15,300万人を超え、死者数は330万人に達する勢いである。国内でも市中感染が広がり、第4波に突入したと考えられている。各国で治療薬開発とワクチン開発は熾烈な競争下にあり、国内ではエボラ出血熱治療薬のレムデジビルと抗炎症薬のデキサメタゾンが特例的に承認されているものの、より効果的な治療薬の承認には至っていない。国内感染者への供給の観点から、国産の治療薬やワクチンが強く望まれている。治療薬開発については新規に開発するのでは時間がかかりすぎるため、ドラッグリポジショニングやドラッグリユージングの観点から、既存医薬品から見出すことが主体となっている。本世界初大規模仮想スクリーニングは、2003年のSARSですでに実績のあるメインプロテアーゼ(Mpro)ダイマーを創薬ターゲットとして実施した。メインプロテアーゼはウイルス複製増殖に必須のシステインプロテアーゼで、SARS-CoV-2メインプロテアーゼダイマーのX線結晶構造は2020年3月4日にPDBで公開されたものを用いた。論文は3月31日に投稿しており、わずか1か月足らずのうちに、モデリング、シミュレーション、解析、執筆を行った。著者の開発した構造ベース創薬システムを用いて精密モデリングを実施し、仮想スクリーニングは本システムが搭載するrDockとAutoDock Vinaのインターフェイスを用いて分散処理にて行った。化合物データベースとしてはChEMBL約200万化合物を用い、このうち、開発段階の医薬品から承認薬を13,000個以上含んでいるデータベースである。ChEMBLデータベースを用いた理由は、すべての化合物が文献既知であり、合成可能であるという点である。ドラッグリポジショニングの観点から、まず、rDockで一次スクリーニングを実施し、約100分の1、つまり、2万化合物程度まで絞り込んで、64品目の医薬品を含むヒット化合物を得た。これらは、抗菌剤、抗生物質、抗糖尿病薬、抗エストロゲン薬、抗感染症薬、抗炎症薬(NSAIDs)、抗腫瘍薬、心血管疾患薬、胃腸薬、抗エイズ薬、抗精神薬など様々な疾患に対する医薬品であった。この中には、ドイツ霊長類センターの発表したカモスタットや後に東京大学医科学研究所で発表されたフサンと同じスタット系抗炎症薬のセピモスタットや抗エイズウイルス薬として臨床段階にあるクルクミンなども含まれていた。rDockから得られたドッキングモードを初期構造として、さらにドッキング精度を高めたAutoDock Vinaで二次スクリーニングを実施し、最終的に、28品目の生物活性化合物と、加えて、安全性が確認されている承認薬の低用量経口投与睡眠導入剤のエスゾピクロンがメインプロテアーゼに対して強い親和性を示すことが予測された。本論文は、in pressの段階ですでにFEBS Open Bio誌直近5年間のMost Read Articleランキング2位となっており、現在は直近2年間のMost Cited Articleにも選ばれており、FEBS Open Bio誌Award対象論文としてもノミネートされた。
Motonori Tsuji*. Antagonist-Perturbation Mechanism for Activation Function-2 Fixed Motifs: Active Conformation and Docking Mode of Retinoid X Receptor Antagonists. J. Comput. Aided Mol. Des., 31, 577-585, 2017.
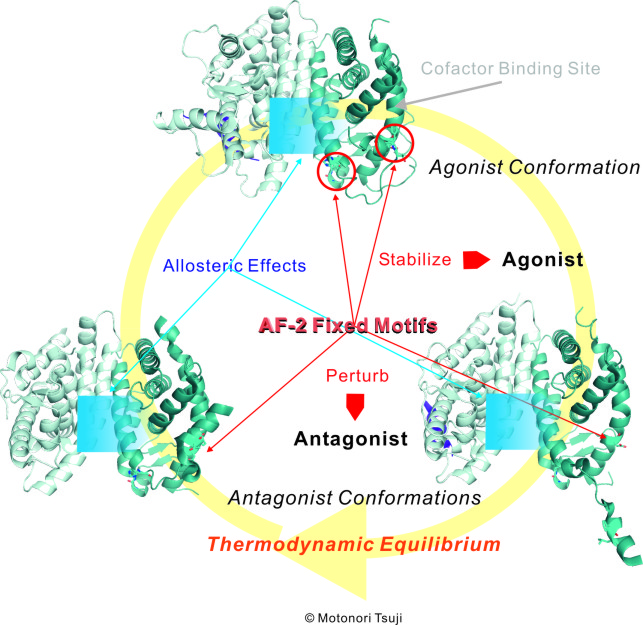
HX531はレチノイドXレセプターアンタゴニスト(RXR)として最初にわれわれが見出した化合物であり、ペルオキシゾーム増殖剤活性化受容体γ(PPARγ)とのヘテロダイマーに作用して脂肪細胞の分化・増殖を制御する抗肥満・抗糖尿病治療薬候補化合物として注目されている。一方、LG100754もRXRアンタゴニストとして最初に合成された化合物である。HX531がRXRホモダイマーおよびRXRとのヘテロダイマーの両者に対してアンタゴニスト活性を示すのに対して、LG100754はRXRホモダイマーに対してのみアンタゴニスト活性を示し、RXRとのヘテロダイマーに対してはアゴニスト活性を示す点でHX531と作用機構が異なる。本研究では、著者が提唱する核内受容体スパーファミリーリガンド結合領域正準フォールドを規定する局所モチーフ理論から予測される、アゴニストによるAF-2固定モチーフ安定化メカニズムとアンタゴニストによるAF-2固定モチーフ摂動メカニズムに基づいて、HX531とLG100754のRXRへの作用機構の違いを明らかにし、局所モチーフ理論の正しさをより強固にするとともに、HX531のリード最適化のための指針を示した。すなわち、著者が独自に開発した生体高分子−リガンドドッキングシミュレーションプログラムを用いた精密ドッキングと量子化学計算による分子シミュレーションからHX531の活性コンフォメーションとドッキングモードを特定したところ、HX531は典型的なAF-2固定モチーフ摂動型アンタゴニストであるのに対し、LG100754はホモダイマーではAF-2固定モチーフ摂動型アンタゴニストであるが、ヘテロダイマーではAF-2固定モチーフ安定化型アゴニストになることがわかった。LG100754がRXRホモダイマーとヘテロダイマーで活性が逆転する理由はダイマーパートナーからのアロステリックな効果に依存しているとする考察で矛盾がなく、レセプターサブタイプ選択性起源の解明に著者が考案した次世代創薬基盤技術としての生体高分子システム全系量子力学計算による分子設計手法がここでも有効であった。
Motonori Tsuji*, Koichi Shudo, Hiroyuki Kagechika. Identifying the Receptor Subtype Selectivity of Retinoid X and Retinoic Acid Receptors via Quantum Mechanics. FEBS Open Bio., 7, 391-396, 2017.
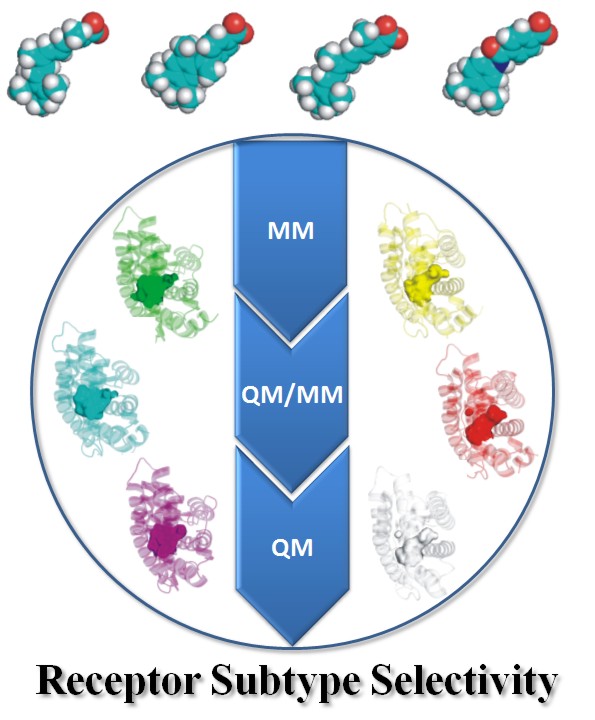
医薬品の標的となるタンパク質(標的タンパク質)の受容体には一般にサブタイプと呼ばれる亜型が存在する。通常、サブタイプは細胞、器官、組織や臓器によって分布が異なっており、その細胞や組織に固有の生命活動に関わっている。サブタイプ選択性を欠く医薬品は全身の細胞や組織に同等に効果を示すため、副作用の原因の一つとなっている。
現在、医薬品はタンパク質の立体構造に基づいて設計(構造ベース創薬と呼ばれる)されており、サブタイプ選択性は医薬品が作用するサブタイプ間のポケットの特徴(形状や物理化学的性質)に依存していると考えられている。そのため、サブタイプ別にポケットの特徴がわかれば、有効な医薬品が設計できると考えられている。この考えに基づいて、医薬品とタンパク質をコンピューター上でドッキングさせるドッキングスタディーやインシリコスクリーニングと呼ばれる方法が最先端の手法として用いられている。これら既存の手法では標的タンパク質のポケット周辺の構造さえわかれば、全体の構造がわかっていなくても医薬品の設計が可能と考えており、計算資源の問題もあり、ポケット周辺の構造情報だけでドッキングを実施している。
今回の研究では、複数の核内受容体と既知医薬品を対象とした全系量子力学計算を実施することで、医薬品の効果が受容体の全体構造に依存していることを明らかにした。その結果、サブタイプ間でポケット周辺の構造に区別がない場合でも、サブタイプ間への選択性あるいは結合親和性をコントロールした医薬品の分子設計が可能であることを示した。
また、医薬品の標的となるタンパク質は、通常、単独では存在しておらず、細胞内の場所や発現時期に応じて別の生体分子と結びついており、こうした別の生体分子からのアロステリックな影響によっても医薬品の効果(サブタイプ選択性や結合親和性の強弱)が変ってくることを初めて量子論的に示した。すなわち、構造ベース創薬に全系量子力学計算を適用することで、既存のドッキング手法では難しかったサブタイプ選択的あるいは組織選択的医薬品の分子設計や探索が可能になることを初めて示した。
今回の研究成果は、サブタイプ選択的医薬品や細胞あるいは組織選択的医薬品を開発するための次世代の創薬基盤技術のための新たな手法として期待される。
Motonori Tsuji*. A Ligand-Entry Surface of the Nuclear Receptor Superfamily Consists of the Helix H3 of the Ligand-Binding Domain. J. Mol. Graph. Model., 62, 262-275, 2015.
核内受容体はリガンド結合領域でリガンドが結合・解離してAF-2領域のコンフォメーション変化を引き起こし、コファクターのリクルートメントを伴って遺伝子転写を制御しているが、構造変化を引き起こす動作原理についてはほとんど解っていなかった。今回、アポ体の特定位置(ヘリックス3)に、リガンドの形状や大きさを認識する3つの残基からなるリガンドエントリー表面があることを発見した(ヘリックス3三点初期結合仮説)。さらに、分子動力学計算を用いて、リガンドがアポ体のエントリー表面に到達することで、ホロ体に構造遷移するシミュレーションに初めて成功した。本シミュレーションの成功を裏付けるために量子力学計算で検証した結果、リガンドとエントリー表面のアミノ酸残基が相互作用することで、エントリー表面の裏側(レセプター内部)で電荷の再配置が起こり、静電ポテンシャルに摂動を与える。これがドライビングフォースとなりリガンドの誘導とレセプターの構造遷移のきっかけとなることがわかった。つまり、アポ体からホロ体への構造遷移は、誘起効果によってレセプター内部の静電ポテンシャルが再構成される過程であると結論した。結果、リガンド誘導と構造遷移に関わるドライビングフォースを明らかにし、核内受容体のリガンド認識機構とアポ体からホロ体への構造遷移の動作原理を原子・電子レベルで明らかにすることに成功した。
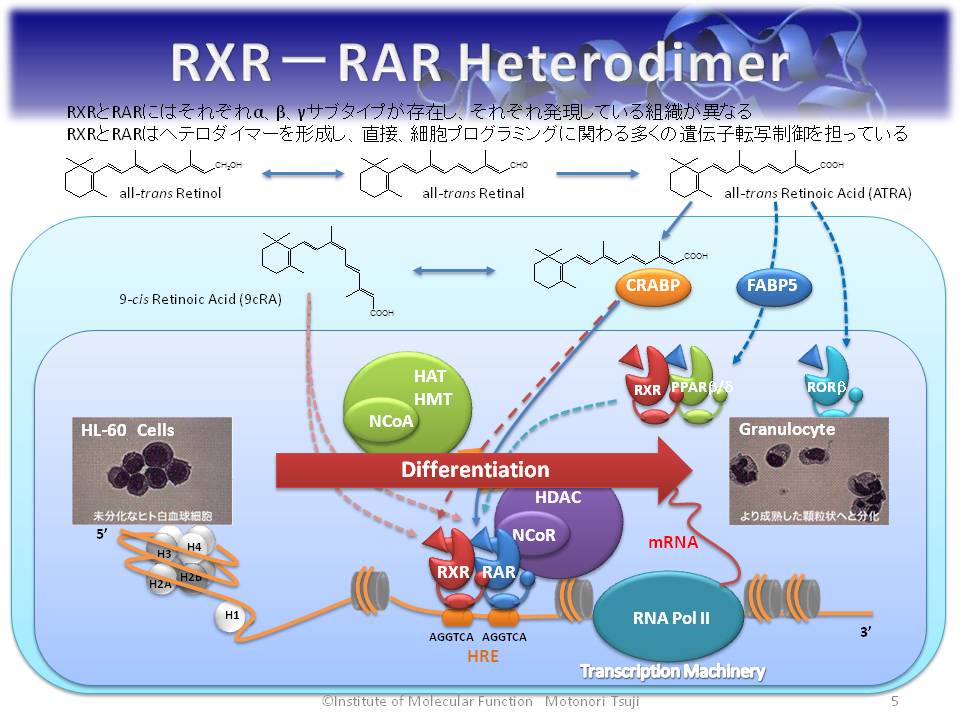
Motonori Tsuji*, Koichi Shudo, Hiroyuki Kagechika. Docking Simulations Suggest that All-trans Retinoic Acid Could Bind to Retinoid X Receptors. J. Comput. Aided Mol. Des., 29, 975-988, 2015.
All-trans レチノイン酸(ATRA)は核内に移行した後、核内受容体であるレチノイン酸レセプター(RARs)と結合する。RARsはレチノイドXレセプター(RXRs)とプロモーター領域にあるDNA上のホルモン応答配列でヘテロダイマーを形成しており、ATRAの結合と解離に応じてリガンド結合領域にあるAF-2領域の大きな構造変化を引き起こし、コファクターのリクルートメントを伴って様々な遺伝子の転写を制御している。In vitroではRXRsはATRAの幾何異性体である9-cis レチノイン酸(9cRA)と強力に結合することがわかっているが(9cRAはRARsにも強力に結合する)、9cRAは胚細胞において検出されない。一方、ATRAは比較的豊富に存在する。レチノイド化学において、RARsの内因性天然リガンドはATRAであり、ATRAはRXRsには結合しないとされている。著者らはATRAがRXRsにも結合でき、そのためにATRAの生理学的な多面的効果が惹起されるとの仮説を立ててきた。本研究では、著者が独自に開発した生体高分子−リガンドドッキングシミュレーションプログラムを用いて精密ドッキングを行い、得られた相互作用エネルギーとわれわれの実験で得られている結合親和性との間に強い相関を見出した。結果、ATRAはRARsのみならずRXRsの内因性リガンドとしても作用するとするRXRsを介した転写制御メカニズムを提唱するに至った。今回の成果はATRAの作用メカニズムに関する定説を覆す研究として注目に値する。
Motonori Tsuji*. Geometrical Dependence of the Highest Occupied Molecular Orbital in Bicyclic Systems: p Facial Stereoselectivity of Bicyclic and Tricyclic Olefins. Asian J. Org. Chem., 4, 659-673, 2015.
これまでに反応面立体選択性に関する多くの理論や仮説が提唱されている。立体的に小さい置換基の側から起こる反応が優先されるクラム則、クラム側に遷移状態の安定性を考慮したフェルキンモデル、それに軌道論を導入したフェルキン−アーンモデル、遷移状態における超共役効果を考慮したシープラックモデルは代表的な反応面選択性モデルとして知られている。しかしながら、これらモデルで説明できない例外的な選択性を示す反応が多く、いずれの理論も単独では統一的説明が不可能であり、反応面立体選択性問題は有機化学の歴史上でも最大級の未解決問題であった。今回著者は、包括的なモデル実験と計算化学・理論有機化学を駆使し、過去に報告されてきたいずれの実験結果とも矛盾しない理論を提唱し、反応面立体選択性問題の本質的原理の解明に成功した。すなわち、計算化学を用いた解析により、分子構造(幾何学)に応じて特定のσ軌道がスルースペース/スルーボンド軌道相互作用によってフロンティア軌道に混成されることを突きとめた。その結果、軌道リハイブリダイゼーションおよび軌道チルティングによってπ軌道の傾きと大きさが試薬が攻撃してくる方向に有利に摂動を受ける。このことが、立体選択性を示す本質的な原理であると結論した。今回提唱した辻モデルは、1981年にノーベル化学賞を受賞した福井謙一博士のフロンティア軌道理論を拡張する発見として注目に値する。辻モデルは基礎有機化学における重要な発見に留まらず、有機合成化学を駆使する医薬や農薬といった立体選択性を試行錯誤して開発される薬の構造を、分子設計の段階で予測可能にするという意味で重要な発見といえる。
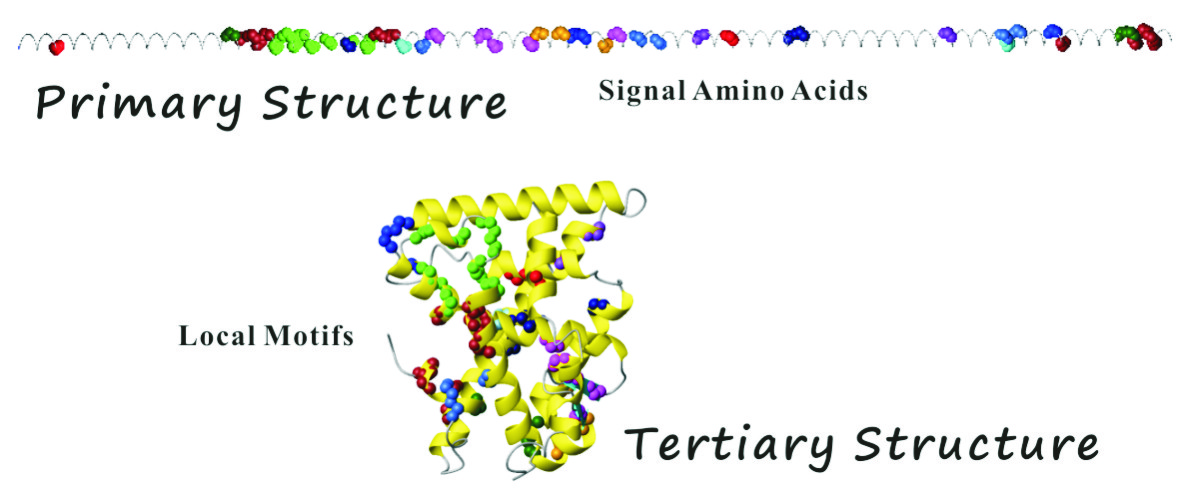
Motonori Tsuji*. Local Motifs Involved in the Canonical Structure of the Ligand-Binding Domain in the Nuclear Receptor Superfamily. J. Struct. Biol., 185, 355-365, 2014.(90日間のダウンロード数が22位を達成しています)
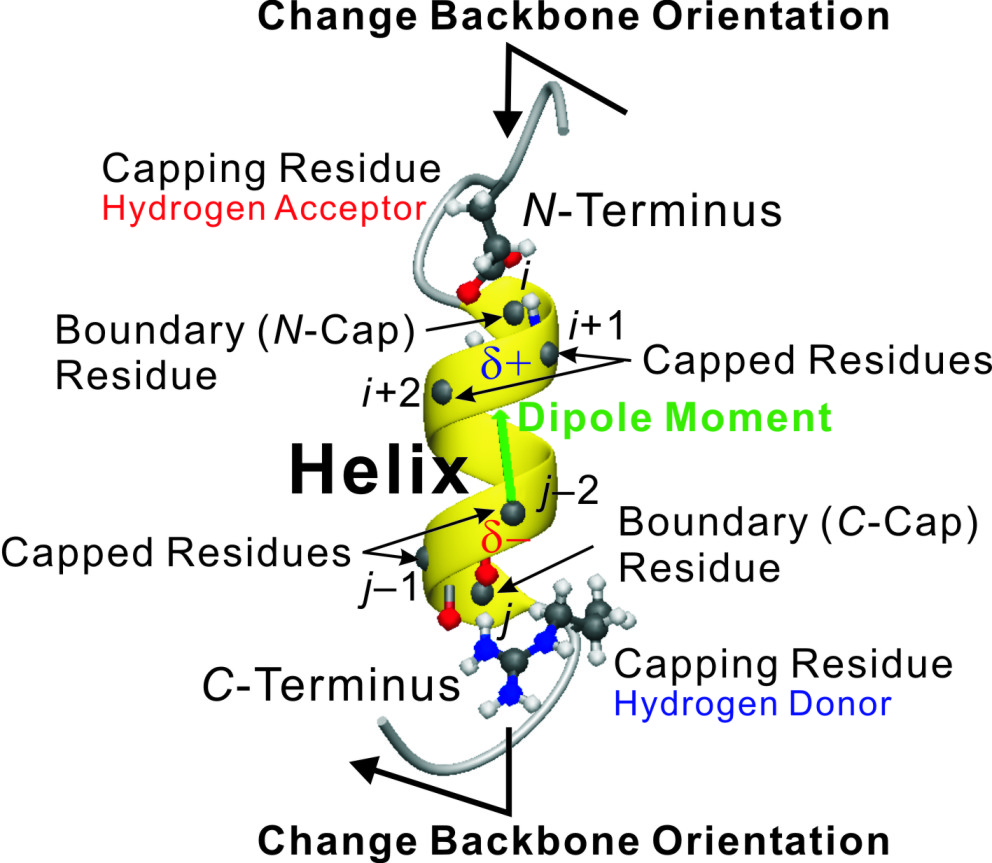
核内受容体スーパーファミリーのリガンド結合領域立体構造が、一次構造で20%以下の相同性でありながらも、例外なく同様のフォールドを採用していること(正準フォールド)に着目し、網羅的なアミノ酸配列および構造アラインメントを実施した。結果、核内受容体リガンド結合領域の正準フォールドとリガンド結合とに密接に関わる極めて重要な相互作用系が少なくとも11ヶ所存在すること見出した。すなわち、正準フォールドを規定しているへリックス、シート、ランダムコイルの始点と終点(境界構造)には進化的に保存されてきた相同アミノ酸残基(シグナルアミノ酸)が位置しており、一次構造上で遠隔位置にありかつ特定の共通位置にあるこれらシグナルアミノ酸が三次構造上で互いに近接して強い相互作用系(局所モチーフ構造)を形成していることを見出した。これら局所モチーフ構造はレセプター内部では疎水環境下での塩橋などの誘電率に依存する静電相互作用、レセプター表面では疎水性相互作用を採用しており、特に、へリックスの始点・終点では電荷―双極子相互作用が関与し、二次構造のオリエンテーションを制御していることを見出した。すなわち、局所モチーフ構造の会合が正準フォールドの決定因子として寄与しているとする核内受容体リガンド結合領域のフォールディング機構を発見し、これを局所モチーフ理論と名づけた。この発見により、アンフィンセンのドグマが核内受容体リガンド結合領域立体構造で成立しており、レビンタールのパラドクスに一つの回答を提示した。さらに、コファクター結合部位、ダイマー形成部位、ホモダイマー・ヘテロダイマー形成タイプの差異、特定アミノ酸残基のポイントミューテーションによってもたらされる立体構造変化などもすべて局所モチーフ理論で説明できることを示した。
核内受容体リガンド結合領域立体構造からアゴニストフォームとアンタゴニストフォームが実験的に確認されており、その主原因がリガンドとの立体反発の有無として解釈されているが、例外も多く、科学的な解明がなされていなかった。しかしながら、局所モチーフ理論に基づいて考察すると、既知リガンドは局所モチーフ構造の相互作用を増強あるいは弱体化する官能基もしくは置換基を有しており、増強する場合にはアゴニスト、弱体化する場合にはアンタゴニストになるとする考察で一貫して説明できることもわかった。すなわち、アゴニズムとアンタゴニズムという現象論に対して、より具体的に電子構造論的に解明し、これを核内受容体におけるアゴニズム・アンタゴニズム理論として提唱した。
さらに、本研究では一次構造上で遠隔位置にありかつ特定の共通位置にあるシグナルアミノ酸をアラインする方法を提案し、既存のマルチプルアランメントとは比較にならない精度の多重アミノ酸配列アラインメント法の可能性を示すことに成功した。
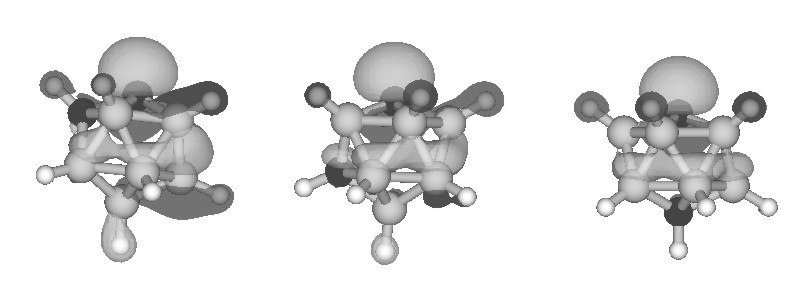
Motonori Tsuji*. Most Stable Conformation of the Cyclopropane Ring Attached at a Carbon Atom in a 1,2-Dicarba-closo-dodecaborane(12) System. J. Org. Chem., 69, 4063-4074, 2004.
C2B10H12の組成式から成るカルボランはほぼ正二十面体の炭素−ホウ素クラスター化合物で、2個の炭素原子の相対配置からオルト、メタ、パラ異性体が存在し、三次元ベンゼンと称される。しかし、カルボランの物性に関しては科学的解明がなされておらず、芳香族性を有しているかについても不明であった。著者はカルボランの電子構造を明らかにする目的で、世界で初めてカルボラニルカルボカチオン生成に関する実験的・理論的研究を行ってきた。特に、オルトカルボラン(oCB)は炭素原子同士が隣接していることから、一方の炭素原子に各種ホモ共役及び超共役置換基を導入することでカルボカチオンの安定化を試みてきた。この研究過程で得られたシクロプロピル-oCBの低温結晶構造は、シクロプロピル基の安定コンフォメーションとして知られているバイセクティット(二等分型)及びパーペンディキュラー(垂直型)コンフォメーションのいずれでもない、第三の最安定コンフォメーション(捻じれ垂直型)を採用していることを発見した。これに対してシクロプロピル基と同じホモ共役置換基を有するフェニル-oCBは二等分型コンフォメーションを採用していた。計算化学及び分子軌道理論による解析を行い、第三番目のシクロプロピル基の最安定コンフォメーションは二種類のホモ共役が効果的に関与した場合に生じ、ホモ共役の強弱でoCBの炭素−炭素結合長も大きく変化することを実験と理論により証明した。
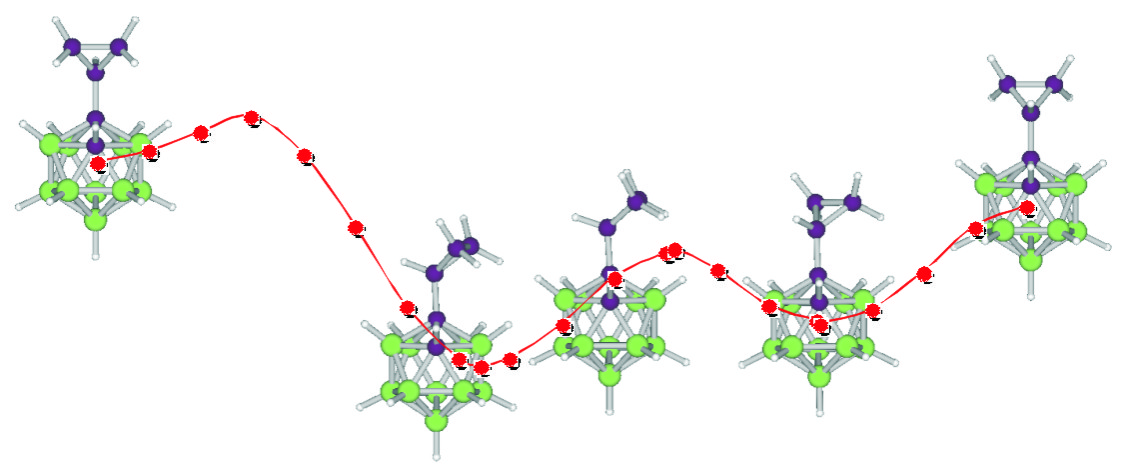
Motonori Tsuji*. On Attempts at Generation of Carboranyl Carbocation. J. Org. Chem., 68, 9589-9597, 2003.
本研究はカルボランをビルディングブロックとした農医薬品開発を主目的として開始した。C2B10H12の組成式から成るカルボランはほぼ正二十面体の炭素−ホウ素クラスター化合物で、2個の炭素原子の相対配置からオルト、メタ、パラ異性体が存在し、三次元ベンゼンと称される。しかし、カルボランの物性に関しては科学的解明がなされておらず、芳香族性を有しているかについても不明であった。著者はカルボランの電子構造を明らかにする目的で、世界で初めてカルボラニルカルボカチオン生成に関する実験的・理論的研究を行った。カルボラニルカルボカチオン存在の根拠として、著者は全異性体のC-ヒドロキシカルボラン(C-ハイドロキシカルボラン、三次元フェノール)をジアゾニウム塩から合成することに世界で初めて成功していた。C-ヒドロキシカルボランの超強酸中での低温NMRではパラ異性体にのみプロトン化が確認でき、カチオンの生成は認められなかった。各種脱離基を導入した加溶媒和反応による反応速度論試験でもC-O結合の解離は認められなかった。そこで、半世紀にもわたったフェニルカチオン存在に関する実験的証明戦略に基づいた検討を試みた。すなわち、オルトカルボランは炭素原子同士が隣接していることから、一方の炭素原子に各種ホモ共役及び超共役置換基を導入することでカルボカチオンの安定化を図った。得られた各種誘導体に対する種々の加溶媒和反応を試みたが、やはりカルボラニルカルボカチオン生成の証拠となる結果は得られなかった。以上の結果から、カルボランは脂肪族炭化水素とも芳香族ベンゼンとも異なる物性を示すと結論した。本研究では、カルボラニルカルボカチオン生成に対する化学的証拠を見出すことはできなかったが、電子的に興味のある様々なカルボラン誘導体を合成し、カルボランの物性に関わる多くの分析結果を得たことで、カルボラン化学に大きく貢献した。
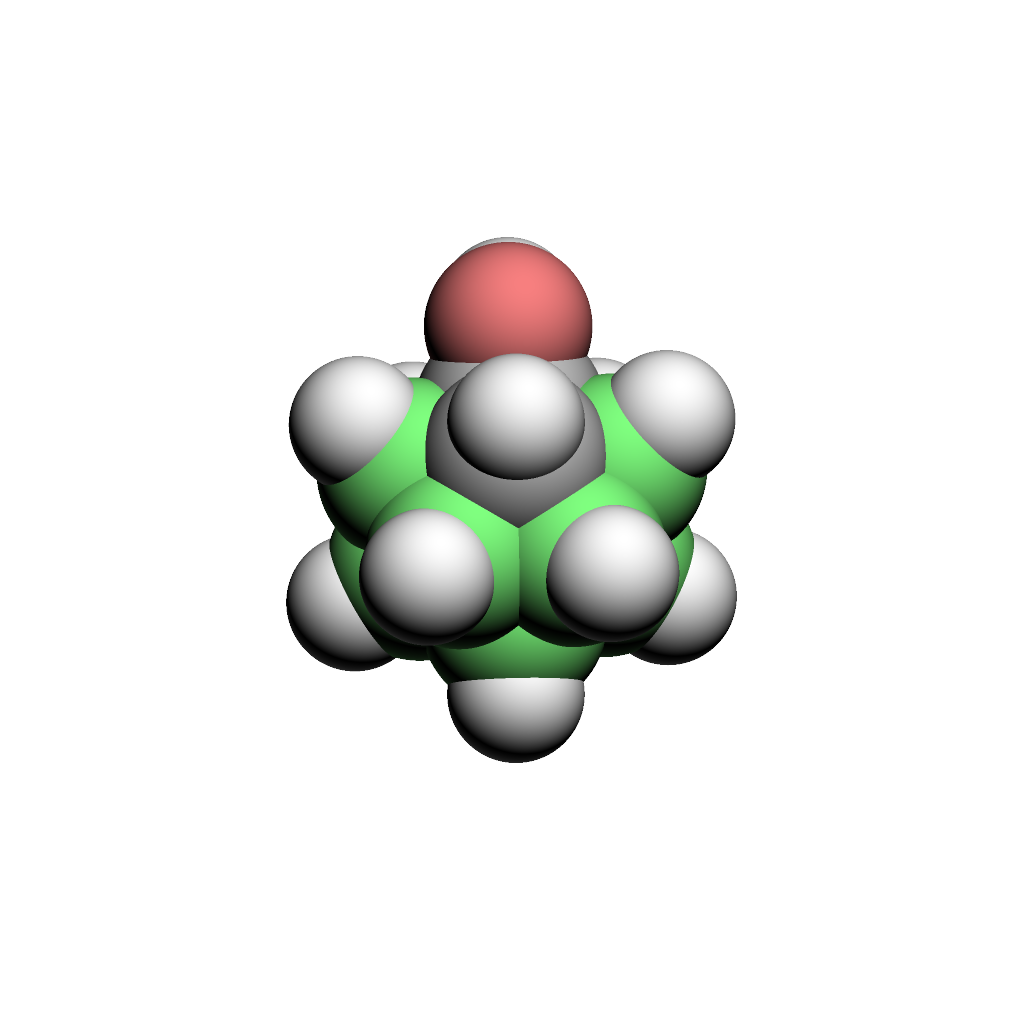
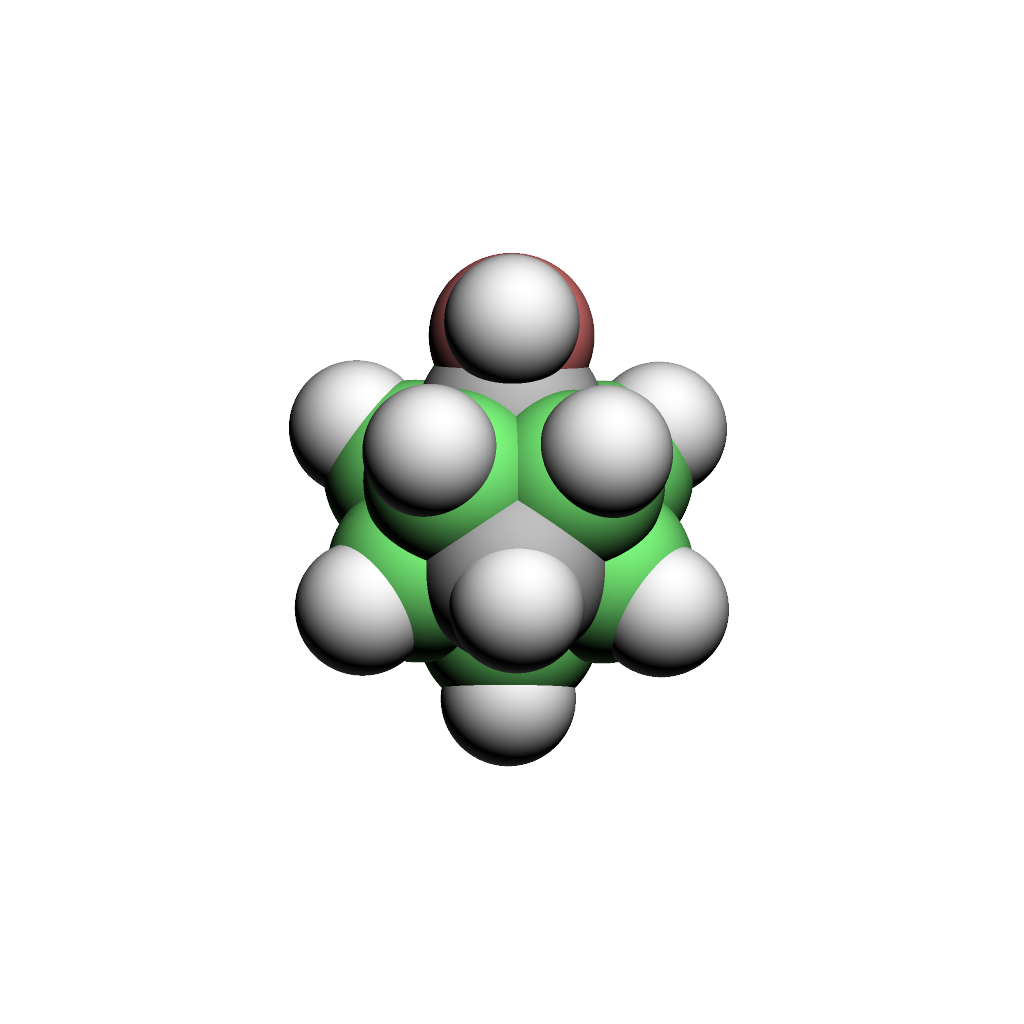
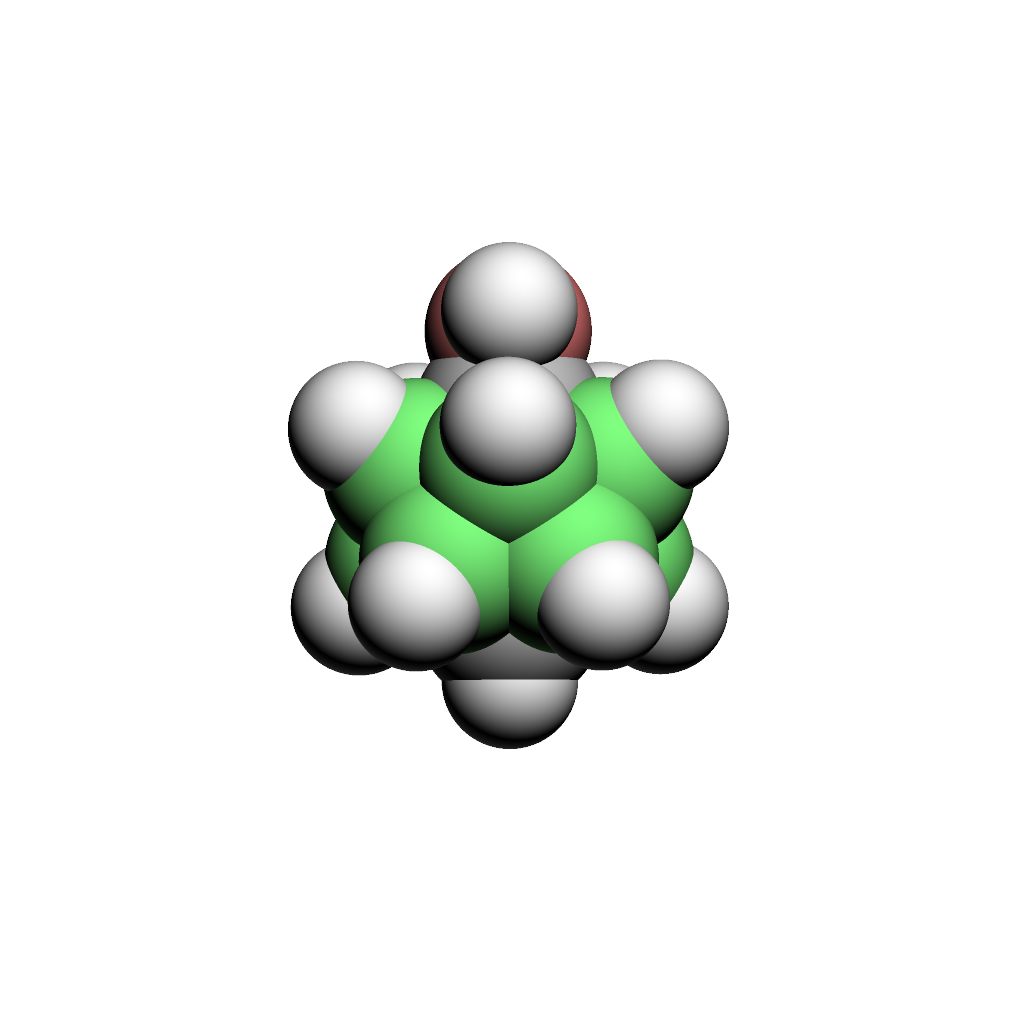
ヒドロキシカルボラン(世界で初めてオルト、メタ、パラ全異性体のヒドロキシカルボラン(C2B10OH12)を求核置換反応で合成に成功(1999年)、現在は様々な医薬品、農薬や機能性材料のビルディングブロックとして利用されている)
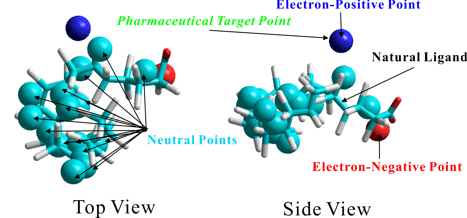
辻一徳.* 生体高分子における相互作用部位の予測方法、出願番号:特許出願2006-125188、公開番号:特許公開2007-299125、登録番号:第5142179号、登録日:2012年11月30日.
構造ベース創薬では標的生体高分子の立体構造、厳密には安定立体構造の原子座標が必須であり、これは現在、X線結晶構造解析、核磁気共鳴(NMR)または科学的根拠をもったホモロジーモデリング等の分子モデリングで得られる。しかしながら、生体高分子の立体構造は、教科書等で表現される明確な「鍵と鍵穴」の形状を持っている訳ではなく、外観からはどの位置が相互作用部位または複合体形成部位であるか一切判断できない。現在のところ、X線結晶構造解析の技術を用いることによってリガンドの複合体形成部位(すなわち、リガンド結合部位)および高分子等の複合体形成部位を特定できるが、得られた立体構造中にリガンド等が複合体を形成しているかどうかは偶然に頼らざるを得ず、リガンド結合部位あるいは高分子等の複合体形成部位の特定に至らない場合が大部分である。その他の技術として、ポイントミューテーションや放射線標識といった化学的手法やNMRや蛍光などの分光学的手法が存在するが、いずれも膨大な実験と労力を必要とする一方で、成功した場合であっても非常に広範囲にわたる大まかな領域を推定できる程度の結果しか得られない。
一方、リガンド結合部位や高分子との複合体形成部位を予測するアルゴリズムもわずかであるが知られている。例えばリガンド結合部位を予測するアルゴリズムに関して言えば、これらはいずれもプローブ原子やプローブ分子を用いて、必要処理を施した生体高分子全体あるいは一部を一定間隔の立方体の集合(すなわち、グリッド)で分割して、各格子点上に該プローブを配置して相互作用エネルギーを見積もる方法(すなわち、グリッドベース)または非グリッドベースの手法である。プローブ原子やプローブ分子を用いたこれら手法は、多くの問題を含んでいる。また、高分子との複合体形成部位に関しては、現在知られるアルゴリズムは本質的に生体高分子の疎水的な表面を探索して推定する方法程度でしかない。上記の問題点を克服する方法については、現在、国家プロジェクトとして、全世界で様々な方面から巨額を投じ、しのぎを削って研究されているが、有効な手法は知られていない。
ドッキングシミュレーションやバーチャルスクリーニング等の構造ベース創薬は、いずれもリガンド結合部位が特定された後に使用される技術であり、リガンド結合部位または高分子との複合体形成部位が不明な大部分の生体高分子については、これらの技術を適用できないのは言うまでもない。逆に、リガンド結合部位または高分子との複合体形成部位が正確に予測することが可能となれば、その後の様々な応用が期待される。
本発明は、生体高分子における内部遠隔構造あるいは金属原子、低分子や高分子などの外部因子との相互作用部位を探索し、該相互作用部位に親和性のある化学基、化学種または分子種を予測する方法を提供するものである。
本発明者は、生体高分子全体構造を取り扱う既存の手法とは全く異なった視点、すなわち、生体高分子を構成し、化学的性質を損なわない程度まで最小化した基本フラグメントに生体高分子全体構造を分割し、該基本フラグメントから全体構造を再構築することで、高度に複雑化した生体高分子のエネルギー的性質を基本フラグメントにおけるより単純化されたエネルギー的性質に帰属させるという方法により本課題を解決した。すなわち、本発明者は、生体高分子構成原子の相互作用点情報をデータベース化し、前記データベースを参照して、任意の生体高分子構成原子の原子座標情報から相互作用点の位置(座標)を算出することによって、前記生体高分子の相互作用部位を予測する方法を開発することに成功した。