![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
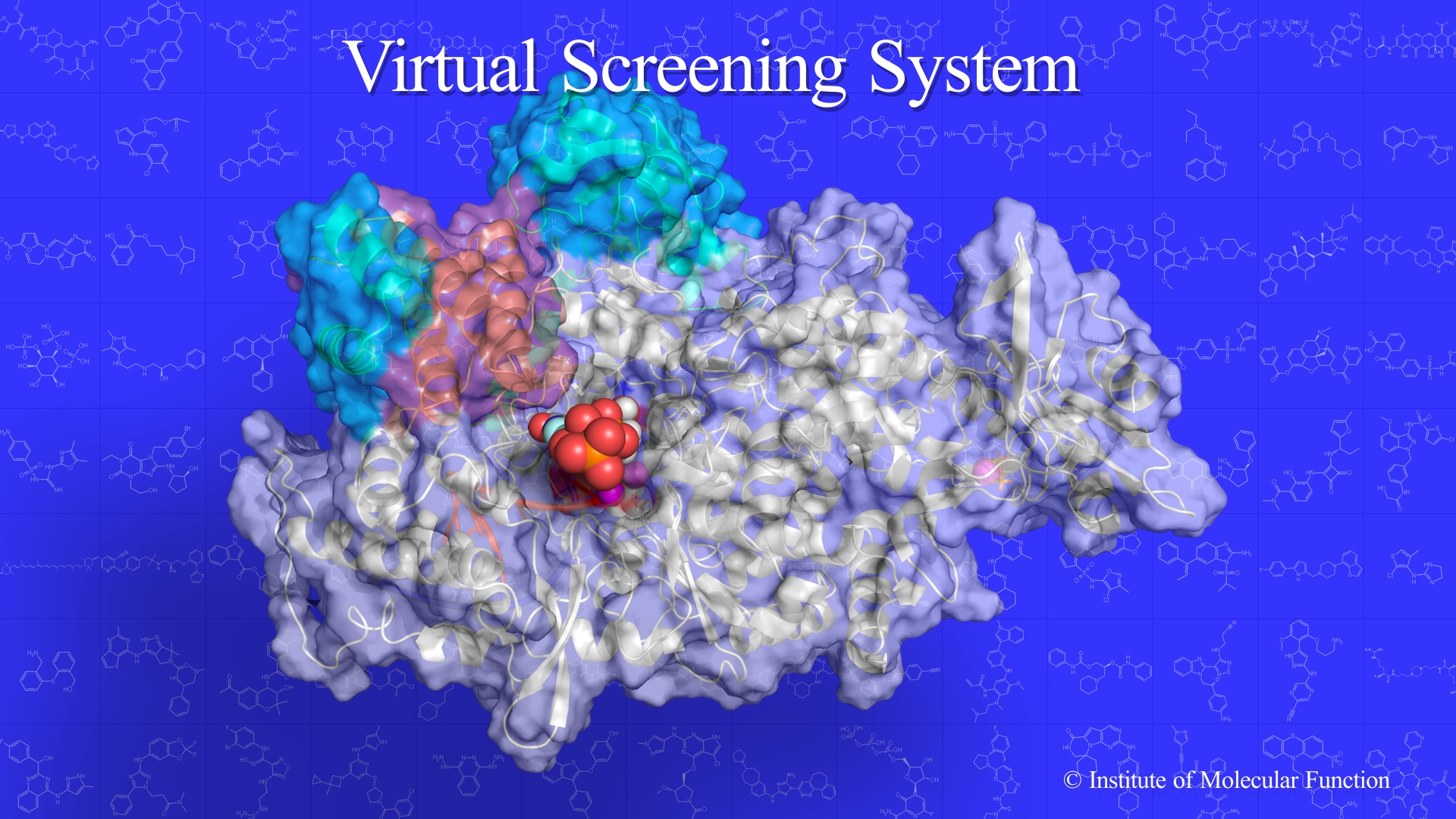
Virtual Screening
Virtual screening is an integrated technique in which the docking algorithm is the core technology.
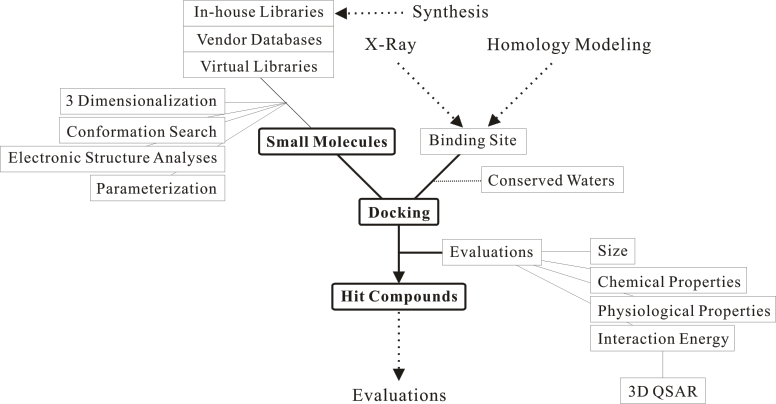
Click on a key word to move to the detailed page.
Virtual Screening System is the latest structure-based drug design system.
Advices for performing the Structure-based Drug Design
In order to obtain a structure-activity relationship, the 3D structure of a target protein is modified by using the 3D structures of the known active compounds.
Moreover, some different models of this protein must be prepared for an effective screening.
When water molecules are observed in the substrate binding site of the related proteins, these water molecules will be able to use for the justification of a model (see the Estimation of Hydrogen Atoms in Water program).
Many initial structures of a single compound must be prepared, even though the compound-flexible docking is used. See the Docking Algorithm.
An useful docking program will be able to hit a lot of compounds with which chemical groups differ each other.
As a result, these candidates can be selected to (or reduced in) a practical amount by using some thresholds which are based on experience rules rather than chemical basis.
A compound library is more important element than a target protein 3D model in a structure-based drug discovery. That is, the library needs to contain active compound to an evaluation system.
Moreover, the active conformation for the target protein needs to be contained in the prepared initial structures of the compound, even though the docking mode is estimated correctly (see Docking Algorithm).
See the following terms for details.
May 2005