

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
Homology Modeling Professional for HyperChem
The procedure and the parameter settings in the individual module programs are shown in the attached manual in detail. The HyperChem operations such as the parameter settings of the geometry optimizations and molecular dynamics annealings are also shown in the manual in detail.
Homology Modeling Tutorial (Globular Protein: human Mineralocorticoid Receptor Ligand-Binding Domain)
Program: Homology Modeling Professional for HyperChem Revision B1, 2007
1. Initiates the "Control Center" by double-clicking on the "HM" icon.
2. Loads the structure of the human glucocorticoid receptor ligand-binding domain as a template into the HyperChem workspace.
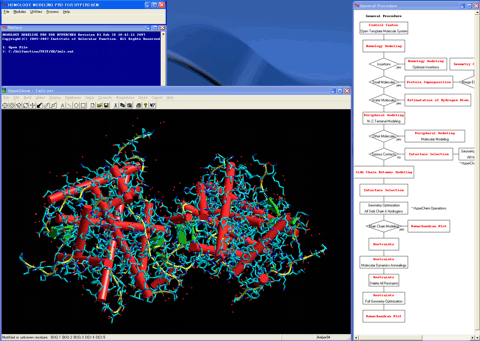
3. Starts the "Homology Modeling" module program.
4. Specifies a sequence of the human mineralocorticoid receptor as a target sequence.
A calculated alignment table will appear (The program automatically performs a pairwise alignment using the Blosum62 score matrix in default, and assigns the secondary structure parts for the template sequence.).
5. Decides a suitable template molecule from the protein molecules which formed the homo-dimer, comparing their respective thermal factors and amino-acid identities with the target sequence.
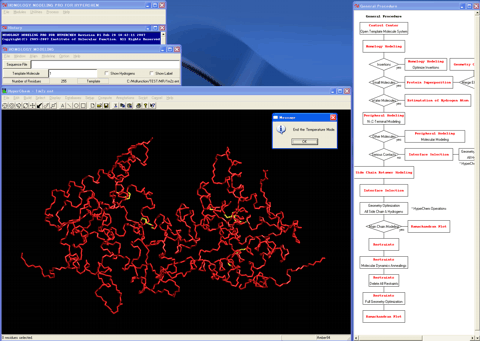
6. Optimizes the alignment if necessary.
7. Assigns structure of an insertion sequence in the target sequence with a secondary structure information obtained from the secondary structure prediction or assigned by a certain method.
8. Assigns protonation state of a histidine residue in the target sequence if necessary.
9. Sets a desired calculation condition for modeling the insertion sequences.
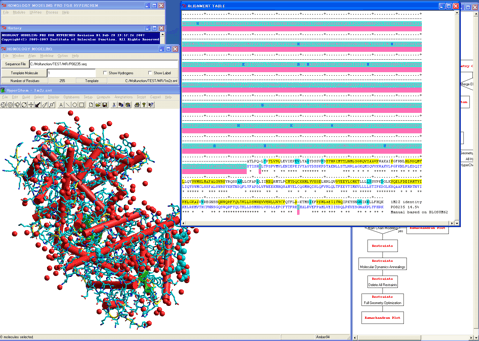
10. Creates a preliminary 3D structure, i.e., model, for the target sequence.
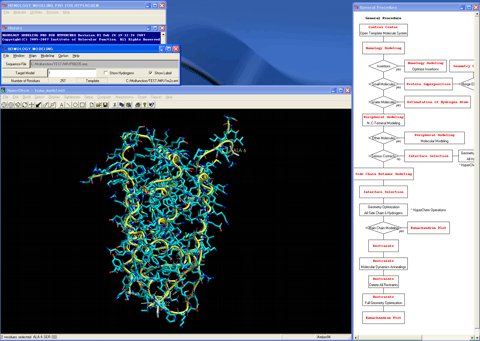
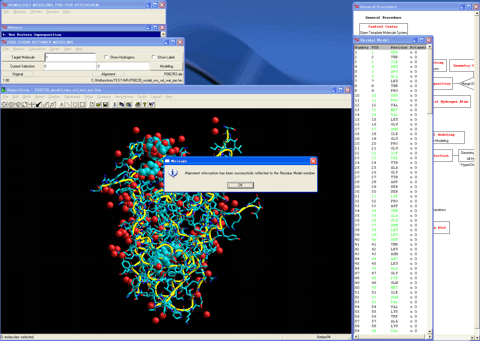
11. Starts the "Protein Superposition" module program.
12. Superposes the structure (blue) of the template molecule system onto the created 3D structure (red).

13. Starts the "Interface Selection" module program.
14. Extracts an important element (contacted to the model) of the template molecule system such as the water molecules, ligand, and co-activator into the created 3D structure.
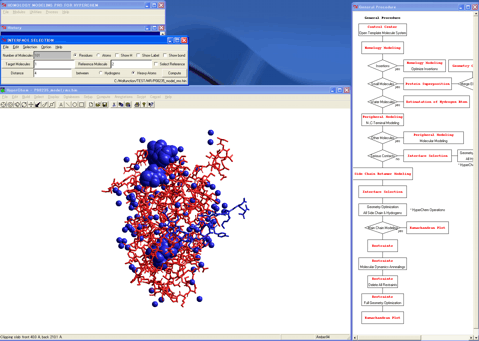
15. Starts the "Estimation of Hydrogen Atoms in Water" module program.
16. Coordinates a reliable initial structure for the hydrogen atoms of the extracted water molecules.

17. Starts the "Peripheral Modeling" module program.
18. Sets an operational molecular mechanics condition.
19. Changes the protonation state of the terminal residues to a zwitterionic state if necessary.
20. Goes to the Small Molecule Modeling mode.
21. Assigns the bonding information, hydrogen atoms, and atom types to the incompleted ligand molecule.
22. Starts the "Gaussian Interface" program.
23. Calculates atomic charges for the ligand molecule using the single point calculations of a suitable theory balanced with the operational molecular mechanics condition.
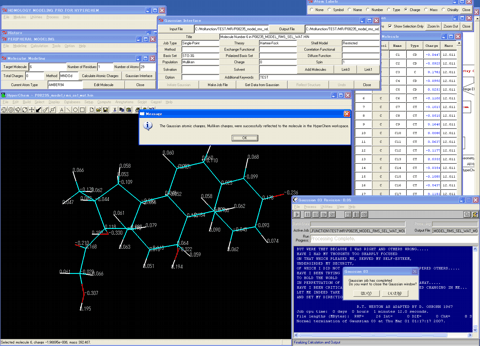
24. Assigns bonding information, hydrogen atoms, and atom types for other molecules if extracted.
25. Starts the "Side Chain Rotamer Modeling" module program.
26. Loads the final alignment table to the program.
27. Sets the operational molecular mechanics conditions and the automatic rotamer search conditions.
28. Determines the best initial rotamer for a residue which differs from the corresponding residue of the template molecule using the batch calculations.
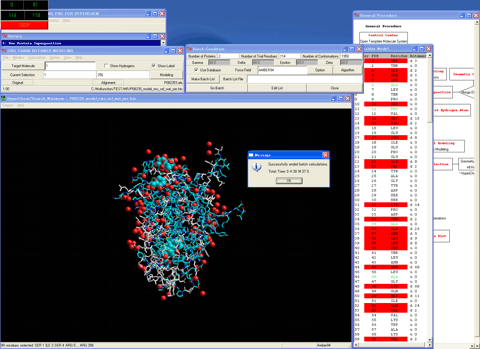
29. Starts the "Interface Selection" module program.
30. Selects the side chain atoms of the protein molecules and all hydrogen atoms of all molecules.
31. Optimizes these atoms under the operational molecular mechanics condition.

32. Starts the "Restraints" module program.
33. Sets the restraint condition to all heavy atoms.

34. Performs the low-temperature molecular dynamics annealings under the operational molecular mechanics condition.
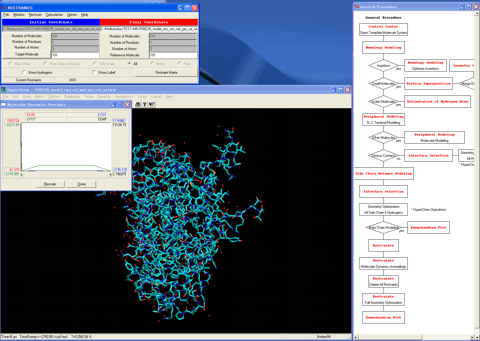
35. Removes all restraint conditions.
36. Performs the full optimizations under the operational molecular mechanics condition.

37. Starts the "Ramachandran Plot" module program.
38. Confirms the precision of the model.
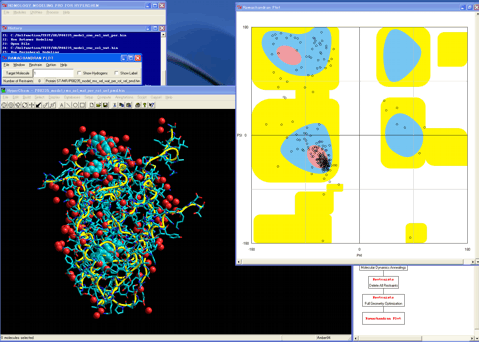
Completed the Homology Modeling for the human mineralocorticoid receptor ligand-binding domain.
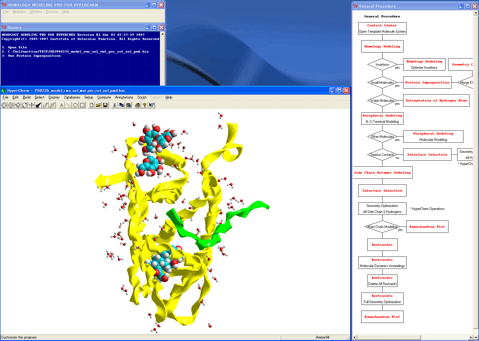
It only takes about two hours (2 h) in our program system, although it takes several days or several weeks in other program systems. In addition, Homology Modeling for HyperChem guarantees the logicalness, reproducibility, and comprehensiveness for the model due to the energy basis (instead of the knowledge basis of other program systems). Moreover, Homology Modeling for HyperChem provides the advanced molecular modeling environment by which the individual researchers can reflect their chemical background to the model, as well.