

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
Structure-based Drug Design
Design of Human Retinoid X Receptor Antagonists and Lead Optimizations
The retinoid X receptor antagonists are the candidate as an effective drug for several diseases in terms of its biological roles. Of the RXR antagonists, HX531 is the well-known, strong RXR antagonist.*
* Chem. Pharm. Bull.,1999, 47, 1778-1786.
Procedure for the Lead Optimizations
Preparations:
The structures of the receptor (including water molecules) and the compound were prepared using Homology Modeling Professional for HyperChem revision B1. The stable conformation of HX531 was obtained from the high-temperature molecular dynamics annealings under the MM+ force field. The structure of HX531 was further optimized at the B3LYP/6-31G*//AM1 model chemistry using Gaussian program via Gaussian Interface for HyperChem. The optimized structure and the Mulliken atomic charges were reflected to HX531 in the HyperChem workspace. The ligand-flexible and the protein- and ligand-flexible docking simulations were performed under the Amber94 force field and the all atom conditions using Docking Study with HyperChem Professional revision A3.
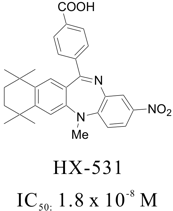
HX531 will be in a rapid equilibrium between each enantiomers arising from the inversion of the nitrogen atom in the benzodiazepine seven-membered ring.
The results of the ligand-flexible docking simulations for these two enantiomers have suggested that the active enantiomer of HX531 has the structure of the left hand side in the above figure, because the other enantiomer of HX531 (right hand side of the above figure) could not form a stable complex in the present docking study.
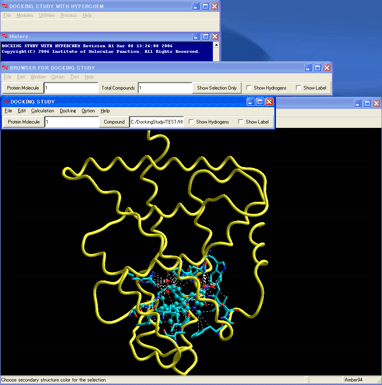
The obtained structure of the stable complex (in the following figure) well agreed with the predicted structure in the literature wherein the first structure of the RXRa ligand-binding domain with the AF2 antagonist conformation of the helix 12 was reported.
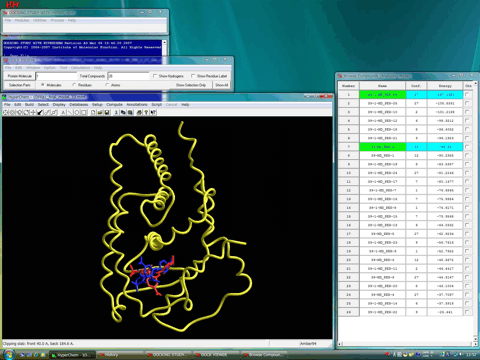
However, the protein- and ligand-flexible docking simulations for these enantiomers suggested that the other enantiomer can adopt a very stable and reasonable complex with the receptor as well as reproduced the stable complex observed in the ligand-flexible docking simulations.
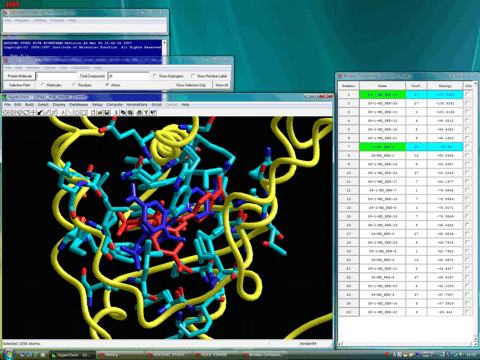
The following figure shows these stable complexes (blue and red).
The following figure shows the flexible parts of the receptor in tube style together with these complexes. A reasonable induced-fit effect of the receptor is observed for both complexes.
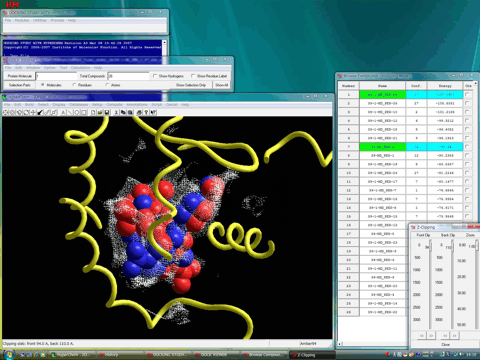
The following figure shows the van der Waals surface of the cavity. Both complexes well fulfill the volume of the cavity.
Although the lead optimizations of HX531 have been investigated by many research groups to date, the biological activity of HX531 has never been improved.
2005/11/06
Return to the Nuclear Receptor Superfamily top page.