

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
Structure-based Drug Design
Design of Human Retinoid X Receptor Agonists
The following table shows the structure-activity relationships of the diphenylamine derivatives, DA series, for the RAR (EC50) and RXR (SEC50) receptors.*
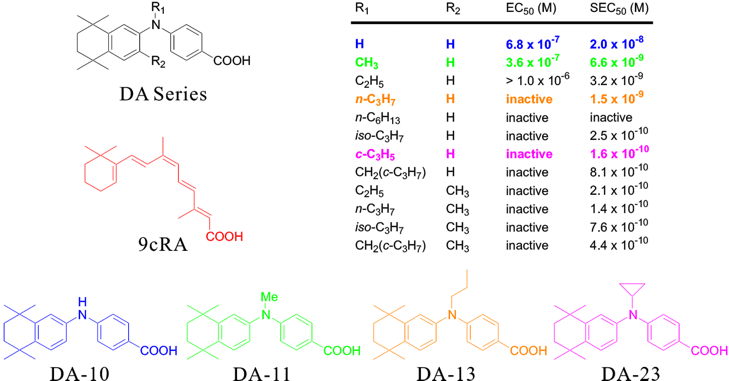
* Biol. Pharm. Bull., 1998, 21, 544-546.
A relatively-low RXR agonist activity as well as a weak RAR agonist activity was observed for the DA10, which has not an alkyl substituent at the N position. When the N-alkyl group is bulkyer, a strong RXR agonist activity with the RXR selectivity was observed. However, larger N-alkyl groups led to loss of agonist activity for both receptors.
Procedure for the Lead Optimizations
Preparations:
The structures of the receptor and the compounds were prepared using Homology Modeling Professional for HyperChem revision B1. The stable conformation of each compound was obtained using the molecular 2D-3D converter, Mol Dimension program, integrated into the Docking Study with HyperChem package. The protein- and ligand-flexible docking simulations for each compound were carried out under the Amber94 force field and the all atom conditions using Docking Study with HyperChem Professional revision A3.
The following figures show the results of the protein- and ligand-flexible docking simulations for some DA compounds, using human RXRα ligand-binding domain with the AF-2 agonist conformation of the helix 12 (including water molecules and co-activator fragment).

The following figure (the table is sorted by the interaction energy) shows the most stable docking mode in terms of the interaction energy of the complex, and simultaneously shows the native ligand, 9-cis retinoic acid (9cRA; red), whose 3D structure of the complex with the human RXRα ligand-binding domain has been solved by X-ray crystal structure analysis. The flexible parts of the receptor are shown in tube style. The resulting structure of the receptor showed the reasonable induced-fit effects. In addition, the interaction energy of the most stable complex for each compound showed an excellent agreement with the observed biological activity.

The following figure shows the van der Waals surface of the cavity, simultaneously.

As the result, all compounds showed the same docking mode as observed for 9cRA.
Moreover, the N-alkyl groups of the DA series corresponded to the methyl group of 9cRA at the 9 position.
In the complex with DA10 (blue), a slight flexibility was observed for the docking mode due to the loss of N-alkyl group. The relatively-low RXR agonist activity of DA10 was accounted in by the weak stabilization of the complex by means of the van der Waals interactions with the hydrophobic amino acid side-chains of the cavity.
In contrast, DA13 (brown) contacted to many hydrophobic side-chains in the cavity. Thus, the strong stabilization of the complex was expected in terms of the van der Waals interactions. On the other hand, the docking mode of this complex slightly differed from those of other complexes. That is, both phenyl rings in DA13 adopted the stacking conformation, whereas both phenyl rings in other compounds adopted the T-shape conformation. This result suggested the presence of some extents of the steric repulsion in the complex with DA13. This consideration consisted with the experimental fact that the n-propyl moiety is a limited size of the N-alkyl group in the DA series for the RXR agonist activity.
Both of DA11 (green) and DA23 (magenta) formed a stable complex with the RXR receptor.
The relatively-low activity of DA11 bearing an N-methyl group was predicted due to reduction of contact with the hydrophobic side-chains in the cavity in comparison with the complex of DA13.
The strongest RXR agonist activity of DA23 in the DA series was predicted, since the bulky N-substituent, cyclopropyl group, led to the formation of the rigid complex with the cavity and no serious contact with the cavity occurred for the complex with DA23 as observed for that with DA13.
From this point of view, we can predict that the introduction of a small hydrophobic substituent of the cyclopropane ring at the 1 position in DA23 will increase the RXR agonist activity.
2005/11/06
Return to the Nuclear Receptor Superfamily top page.