

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
Homology Modeling
Homology Modeling Protocol

Click on a key word to move to the detailed page.
A valuable protein model is given only from the logical homology modeling approach as described below.
Background: Even though a certain amino acid sequence shows only less than 20% homology to the other sequence, there is a case where the corresponding respective 3D structures adopt a single common fold.
In many cases, these proteins constitute a protein family.
In this process, a family or homologous sequence for a target amino acid sequence is searched from sequence databases. Simultaneously, the experimentally-solved 3D structure of this homologous sequence is explored.
In this homology search process, the amino-acid sequences which are suitable as a template can be found from the public databases using FASTA and BLAST methods. Additionally, most of the homology search programs and the sequence databases are available for personal uses.
Alignment of sequences is the most important in homology modeling. That is, only a perfect alignment gives a useful model.
In this process, a preliminary alignment is obtained by the pairwise and multiple alignment analyses, using some score matrices (Blosum and PAM).
See the Homology Modeling program.
Structural alignment is a method that aligns sequences in consideration of the secondary structure parts extracted from the known 3D structures in the family.
The previous preliminary alignment is precisely optimized through this process.
In this process, the additional insertion sequences or the deletion sequences are aligned to those in the loop structures. However, most of alignments obtained from this process are still inaccurate.
See the Homology Modeling program.
Additional chemical logic gives accurate alignment, when applied to this process.
Based on the resulting final alignment, the 3D structure of the target sequence is modeled using the known 3D structure as a template structure.
In general, the side chain conformations (i.e., rotamers) of the corresponding amino acid residues are able to use those of the template protein (see the Rotamer Optimization).
See the Homology Modeling program.
Background: Many insertion and deletion parts of sequence are observed at a loop region.
Background: There is a case that the different proteins, which possess the common amino acid sequence, adopt the same loop structure.
From the fact of these observations, many modeling programs carry the individual loop database.
However, the loop generation method using the loop database is generally restricted to less than 5 residues in the sequence length, since the structure of the same sequence does not find in the structure database. Moreover, the insertions of 20 or 30 length frequently occur in the family sequences.
It has been gradually recognized that a loop structure is also a highly organized structure such as the helix and sheet structures.
In the case of insertions, an inserted sequence frequently constructs an additional secondary structure in the loop.
See the ab initio 3D Modeling program.
See the Homology Modeling program.
Background: A hydrophobic amino acid hardly adopts different rotamers in the core of the protein and on the protein surface. This fact is involved in the property of the interaction system in which the hydrophobic residues can participate.
Background: Polar and charged amino acids hardly adopt the different rotamers in the core of the protein, while the side chains of these residues are flexibly rotated on the protein surface.
Background: It has been known that each of amino acids can adopt some particular side-chain rotamers with a statistical preference in the protein structure.
In other words, modeling of the side chain rotamers must to be performed based on the above chemical basis. Nevertheless, a small difference of rotamer from the true rotamer brings about a large difference of a shape of a ligand binding site.
See the Side Chain Rotamer Modeling program.
All side-chain atoms are optimized using molecular mechanics calculations (e.g., Amber).
Generally, histidine, lysine, and arginine residues are treated as the corresponding cation, and aspartic acid and glutamic acid are treated as the corresponding anion. Moreover, N- and C-terminal residues are treated as a zwitterion.
When hetero molecules such as a ligand are contained in a target system, structural optimization is performed under the presence of these molecules, which must be already treated.
Simultaneously, a hydrogen atom of a hetero atom in the amino acid residue is optimized in consideration of a suitable acceptor.
When the model structure obtained from the target sequence is significantly distorted, the structure will be subjected to the molecular dynamics calculations under the restraint conditions.
See the Peripheral Modeling, the Ramachandran Plot, and the Restraints programs.
May 2005
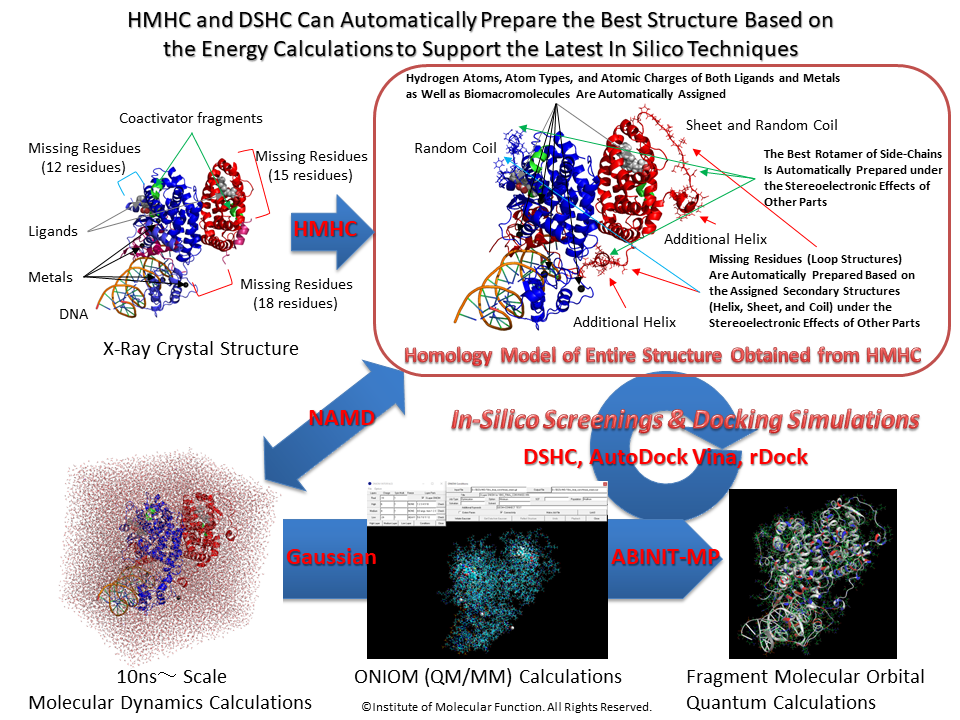
Homology Modeling Professional for HyperChem is a package that consists of many module programs which are required for performing the precise protein homology modeling using HyperChem.
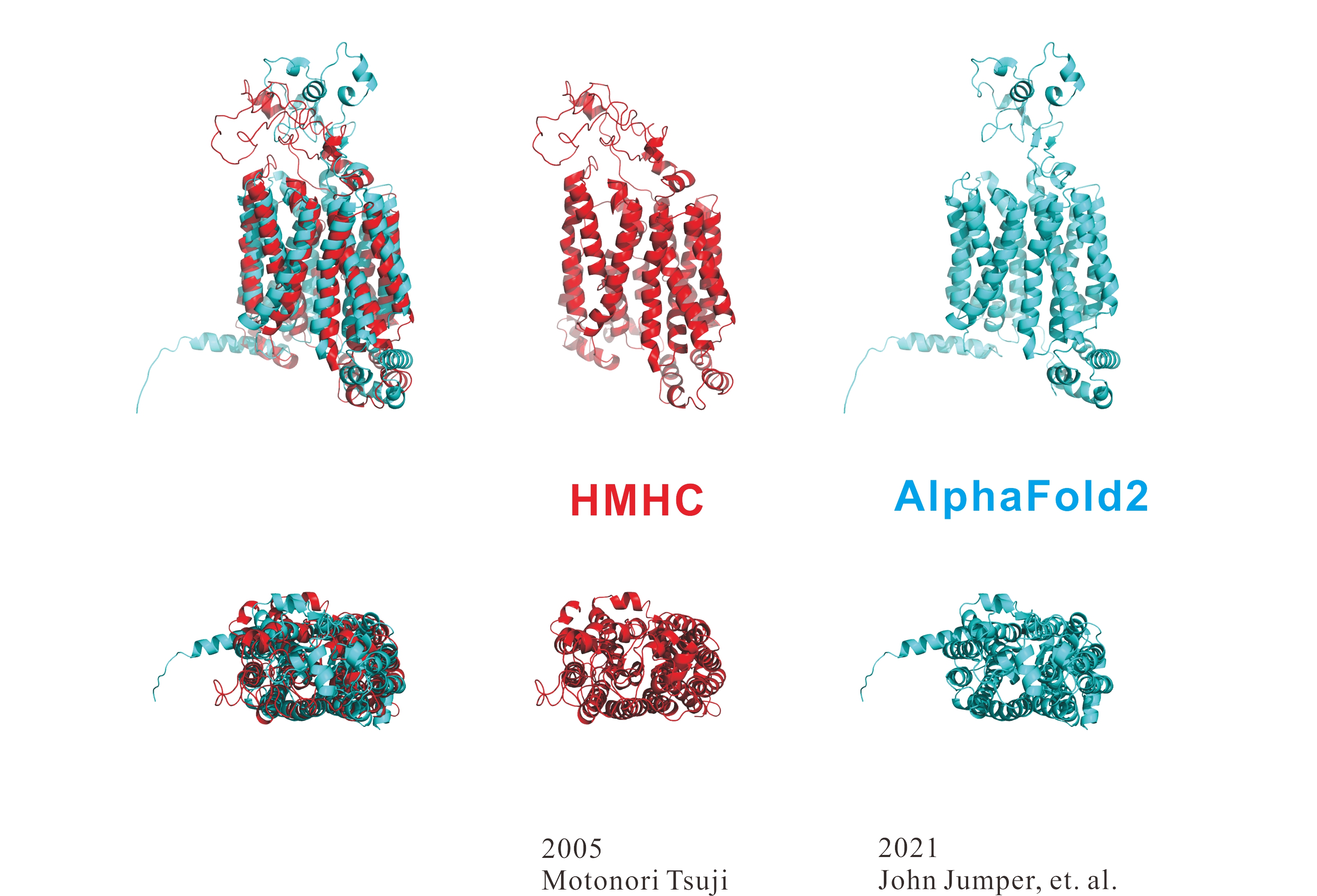
AlphaFold2 vs Homology Modeling Professional for HyperChem (HMHC)
The following figure shows the results of three-dimensional structure prediction for an unknown protein sequence for 20 years including family proteins, using HMHC and AlphaFold2.
HMHC can provide a precise three-dimensional structure since HMHC can model the side-chain rotamer conformations and the interactions between other bioamacromolecules and small molecules as well as crystal waters.
See also Homology Modeling Tutorial.