

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
New Products
What's New in Revision H1. Released the latest version 8.1.5 at 2023/8/10
In Silico Drug Design Total Platform
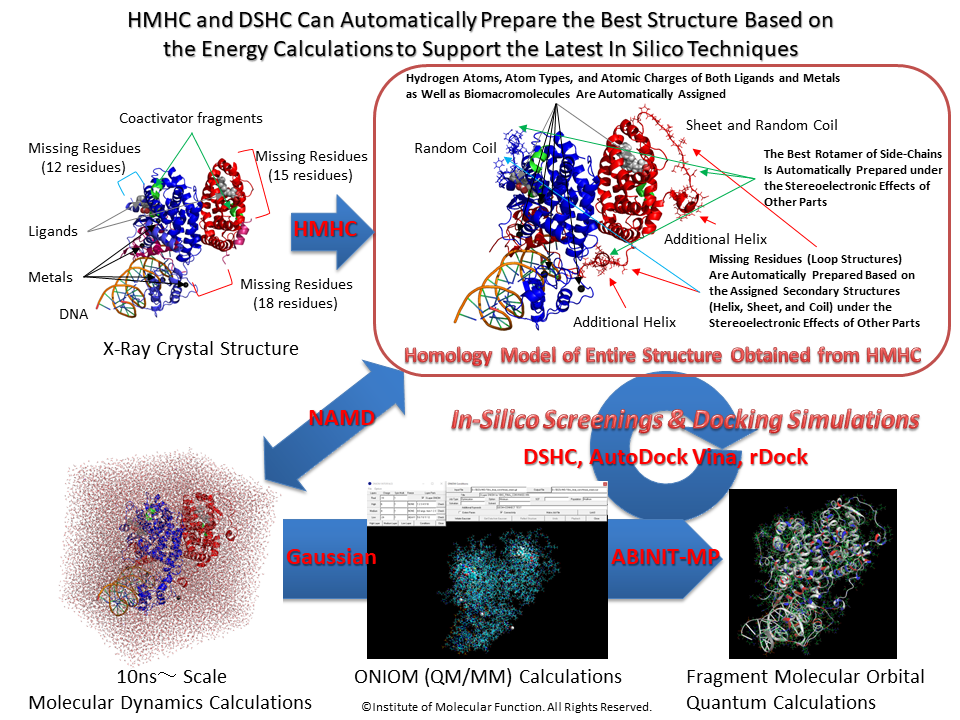
Docking Study with HyperChem
Essential, Premium Essential, Professional, Advanced, Ultimate, and Cluster
with AutoDock Vina In Silico Screenings Interface
Compatible to rDock Docking Simulations, NAMD Molecular Dynamics and ABINIT-MP / GAMESS Fragment Molecular Orbital Quantum Mechanics Calculations for Entire Molecular System
Addition function to the previous revision.
Supported AutoDock Vina Virtual Screenings (unlimited number of compounds independent on package grade) (Preparation of compound database - Preparation of files - Execution - Viewing and Analyzing Vina results - Filtering hits).
Compatible to SDF files obtained from rDock docking programs for analysing docking results, viewing docking modes, filtering hit compounds (DSHC supports stepwise docking simulations using initial structure obtained from rDock followed by AutoDock Vina docking simulations.)
Compatible to file format of fragment molecular orbital calculation programs, ABINIT-MP (BioStation Viewer) and GAMESS (Fu / Facio), for entire biomacromolecular system.
Compatible to CHARMM-based file format of ns scale molecular dynamics calculation program, NAMD (VMD).
Changed default force field to Amber99.
All module programs were improved to keep manual atomic charges of residues in biomacromolecule.
Added the latest manual.
Improved some function.
Manual
Manual Contents (Japanese Language)
Manual 2 Contents (Japanese Language)
Pamphlet
Docking Study with HyperChem, Revision H1 (PDF: 1 MB; Japanese)
Docking Study with HyperChem, Revision G1 (PDF: 2 MB; Japanese)
Homology Modeling Professional for HyperChem
with Gaussian Interface for HyperChem & ONIOM Interface for Receptor
Compatible to NAMD Molecular Dynamics and ABINIT-MP / GAMESS Fragment Molecular Orbital Quantum Mechanics Calculations for Entire Molecular System
Addition function to the previous revision.
ONIOM Interface was largely strengthened for the latestGaussian program.
Compatible to file format of fragment molecular orbital calculation programs, ABINIT-MP (BioStation Viewer) and GAMESS (Fu / Facio), for entire biomacromolecular system.
Compatible to CHARMM-based file format of ns scale molecular dynamics calculation program, NAMD (VMD).
Changed default force field to Amber99.
All module programs were improved to keep manual atomic charges of residues in biomacromolecule.
Added the latest manual.
Improved some function.
Manual
Manual Contents (Japanese Language)
Manual 2 Contents (Japanese Language)
Reference Manual Contents (Japanese Language)
Pamphlet
Homology Modeling Professional for HyperChem, Revision H1 (PDF: 1 MB; Japanese)
Homology Modeling Professional for HyperChem, Revision G1 (PDF: 4 MB; Japanese)
Structure-based Drug Design (SBDD) System
SBDD Pamphlet (PDF: 2 MB; Japanese)
For early revisions:
What's New in Revision G1, 2015/01
What's New in Revision F2, 2012/01
What's New in Revision F1, 2010/02
What's New in Revision E1 and E2, 2008/10
What's New in Revision D1, 2008/05
What's New in Revision C2, 2007/11
What's New in Revision C1, 2007/02