

![]()
![]()
Last Modified
1 January 2024
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
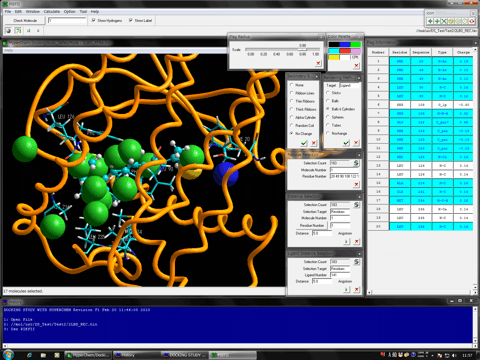
Docking Study with HyperChem
Essential, Premium Essential, Professional, Advanced, Ultimate, and Cluster
What's New in Revision G1.
The full-automatic and comprehensive biological macromolecule (protein and nucleic acid molecule systems)- and ligand (small molecule and peptide)-flexible docking simulations and in silico screenings program.
It's easy to treat the comprehensive conformation search of compound, induced fit effects of the side chain and main chain of the biological macromolecule, and the flexibility of the other molecules such as water molecules, biological molecules, small molecules, and metal atoms, etc.
The program (non-grid algorithm) can predict the precise interaction energy for the entire system rather than the approximated interaction energy obtained from the other docking simulation programs which adopt the grid algorithm for simplifying the energy calculations.
The program adopts the high reliable search method of stable complex using our novel docking algorithm based on the PIEFII technology which can precisely predict the binding site and ligand pharmacophore points in terms of the structure based manner.
In addition to general functions required for the docking simulations and screenings, the flexible docking function in consideration of the induced fit effect of biological macromolecules, the flexible docking function in consideration of large structural changes such as transformation between apo and holo conformations, the flexible docking function under the steric and electronic influences from any other molecules such as the macromolecules, small molecules, water molecules, metal atoms, and substituent groups bound to the target molecule, the atomic charge reassignment function for each conformations using any semi-empirical molecular orbital methods, any combinations of the united atom and all atom conditions, the restart function, the docking function under the solvated conditions, clustering function can be used for performing the precise docking simulations and in silico screenings. Moreover, power users can freely alter any parameters for the docking simulations and screenings as well as those for the molecular mechanics and quantum mechanics calculations via the provided graphical user interfaces.
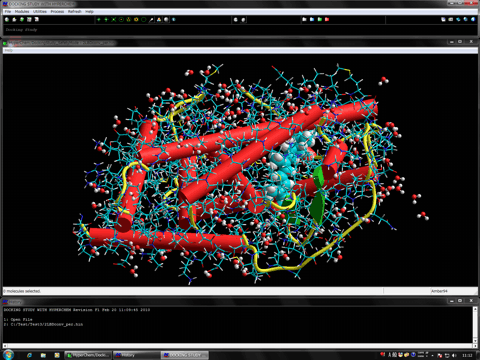
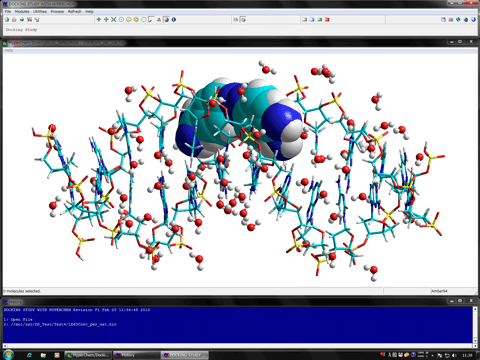
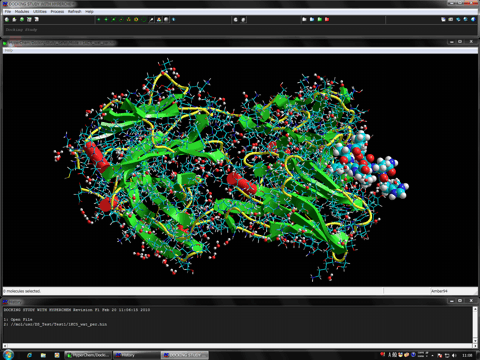
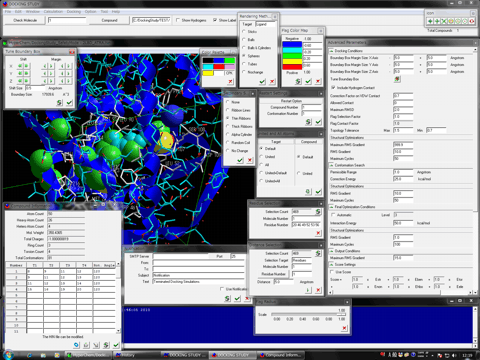
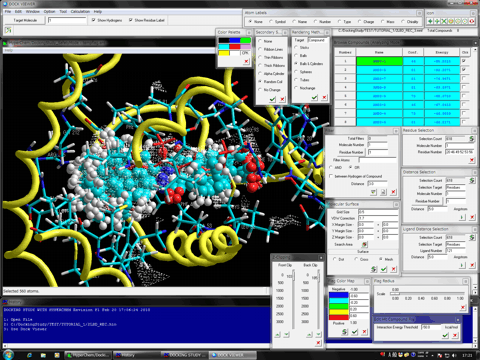
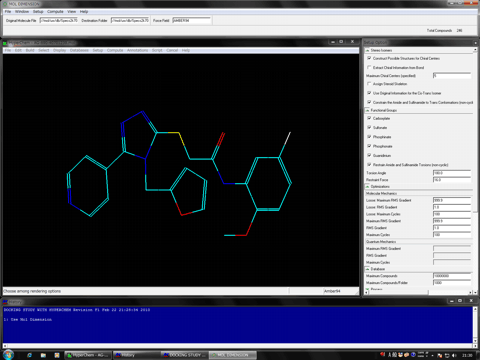
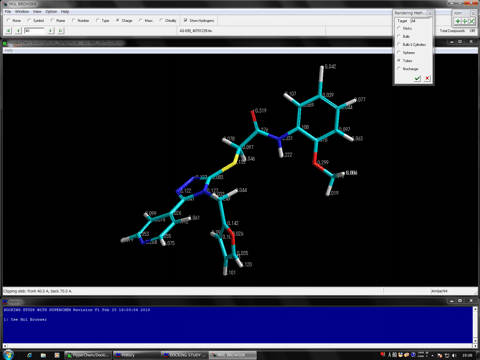
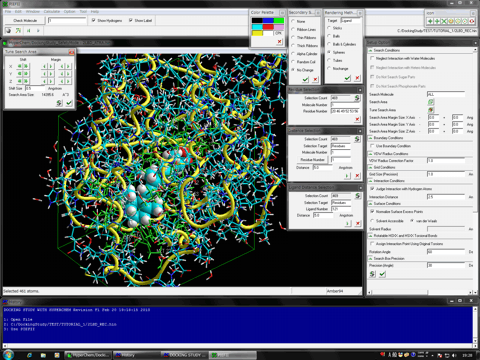
The following shows the addition functions to the previous revision.
The Fast Conformation Search algorithm is available for virtual screenings.
Added structure viewer function to Mol Dimension module program.
Mol Dimension module program is compatible to hypervalent compounds.
Mol Dimension module program is able to separate the big database (100 million compounds) of small molecules in the SDF format into several files of certain size.
Mol Dimension module program can construct the 3D structure database in which the atomic charges and the atomic types of compounds are automatically assigned in the MOL2 format.
Mol Dimension module program can refine the database using rule of five, although this function has previously been available for the Docking Study module program.
RMSD calculation function and table were added to DockViewer module program.
Improved some functions.
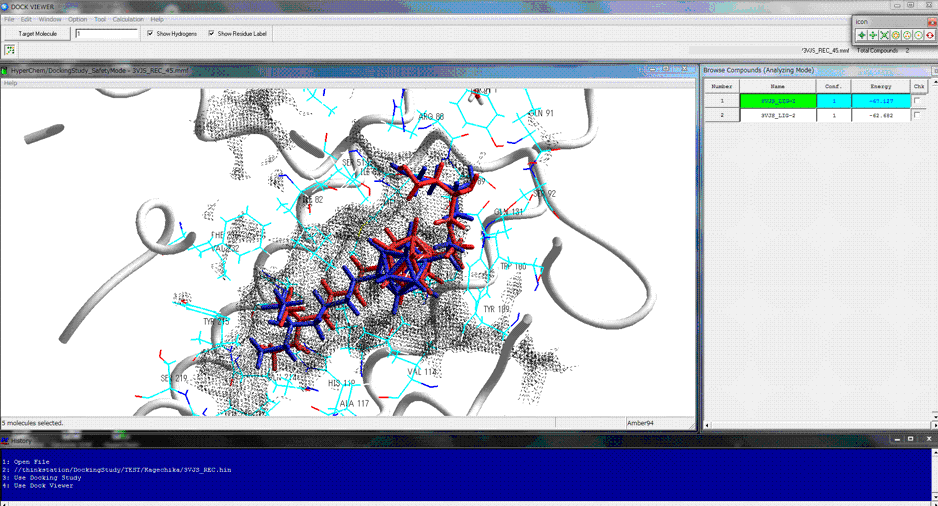
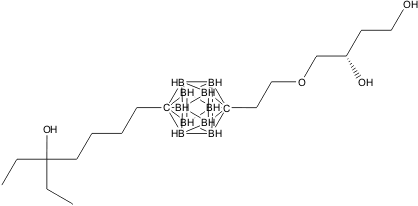
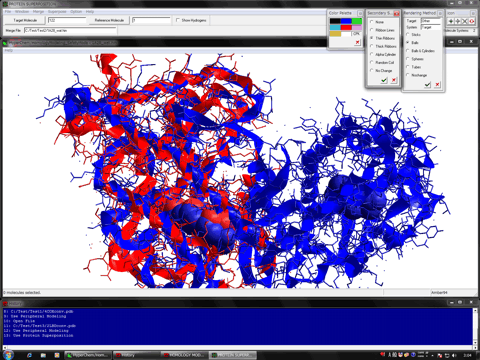
The following figure shows the redocking of the hypervalent compound (carborane derivative). Red tube shows the best docking mode and blue tube shows the crystal structure.
Manual Contents (Japanese Language)
Docking Study with HyperChem (PDF: 2 MB; Japanese)
Homology Modeling Professional for HyperChem
What's New in Revision G1.
Homology Modeling for HyperChem is the latest molecular modeling package which can carry out the molecular modeling, functional analysis, and simulations of a big molecular system using comprehensively the ab-initio quantum chemistry calculations and the molecular dynamics simulations as well as the molecular mechanics calculations.
The program package can be used for performing the protein homology modeling in the presence or absence of the small molecules, water molecules, other biological molecules, metal atoms, and the small molecule covalently bound to the model under the vacuum or the various solvated conditions. The package also provides the several modeling, simulating, and analyzing functions.
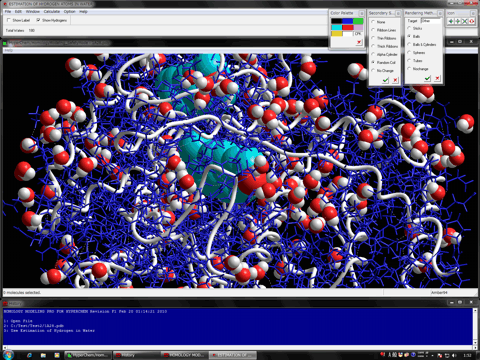
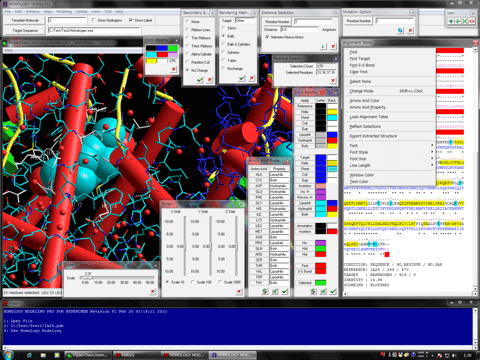
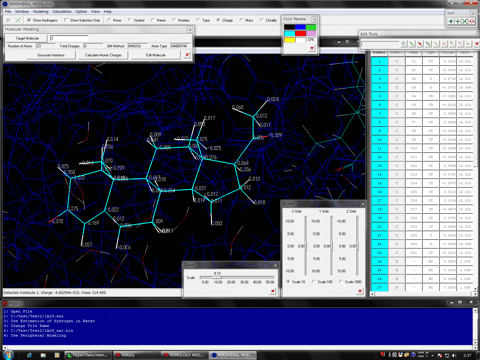
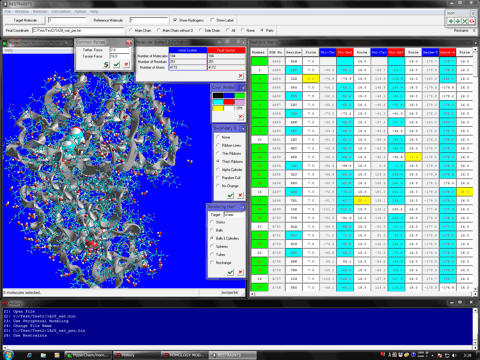
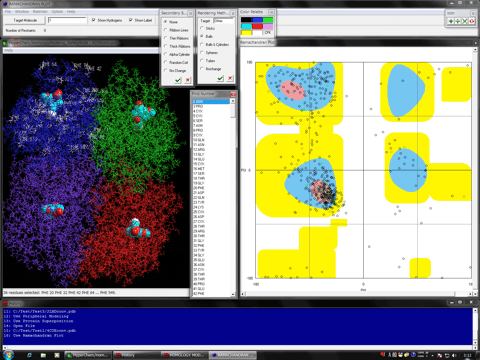
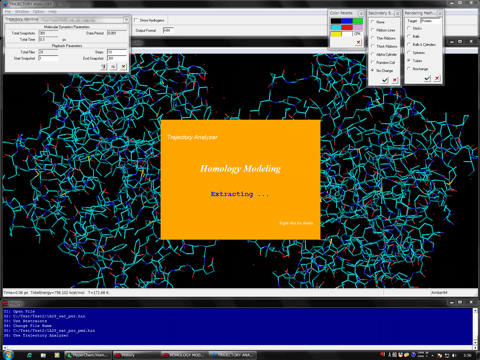
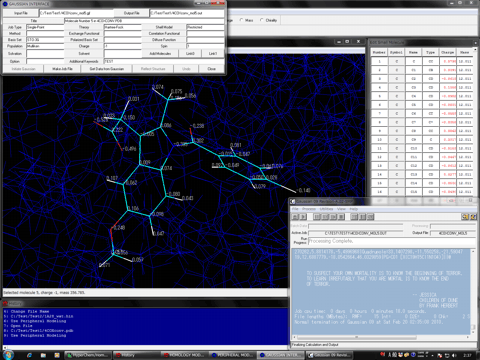

The following shows the addition functions to the previous revision.
Changed the license methods.
The function of ONIOM Interface for Receptor was strengthened (compatible to whole system of protein and nucleic acid molecular system).
Improved the interface of BLAST (new and legacy) program in Control Center module program.
Added the RMSD trajectory (Ca atoms, back bone atoms, all heavy atoms) obtained from MD simulations to Trajectory Analyzer module program.
Improved of some functions.
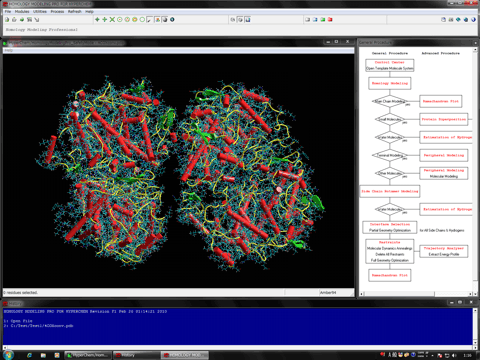
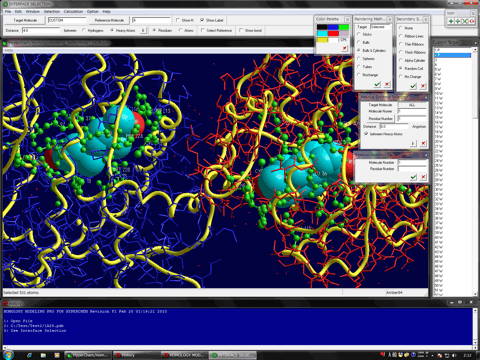
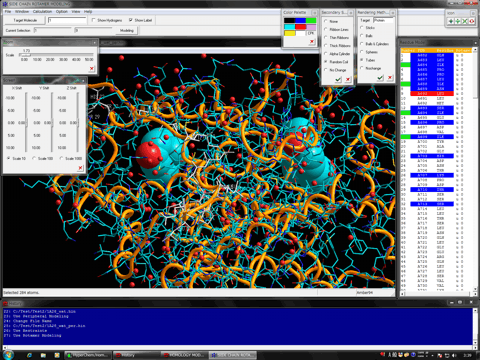
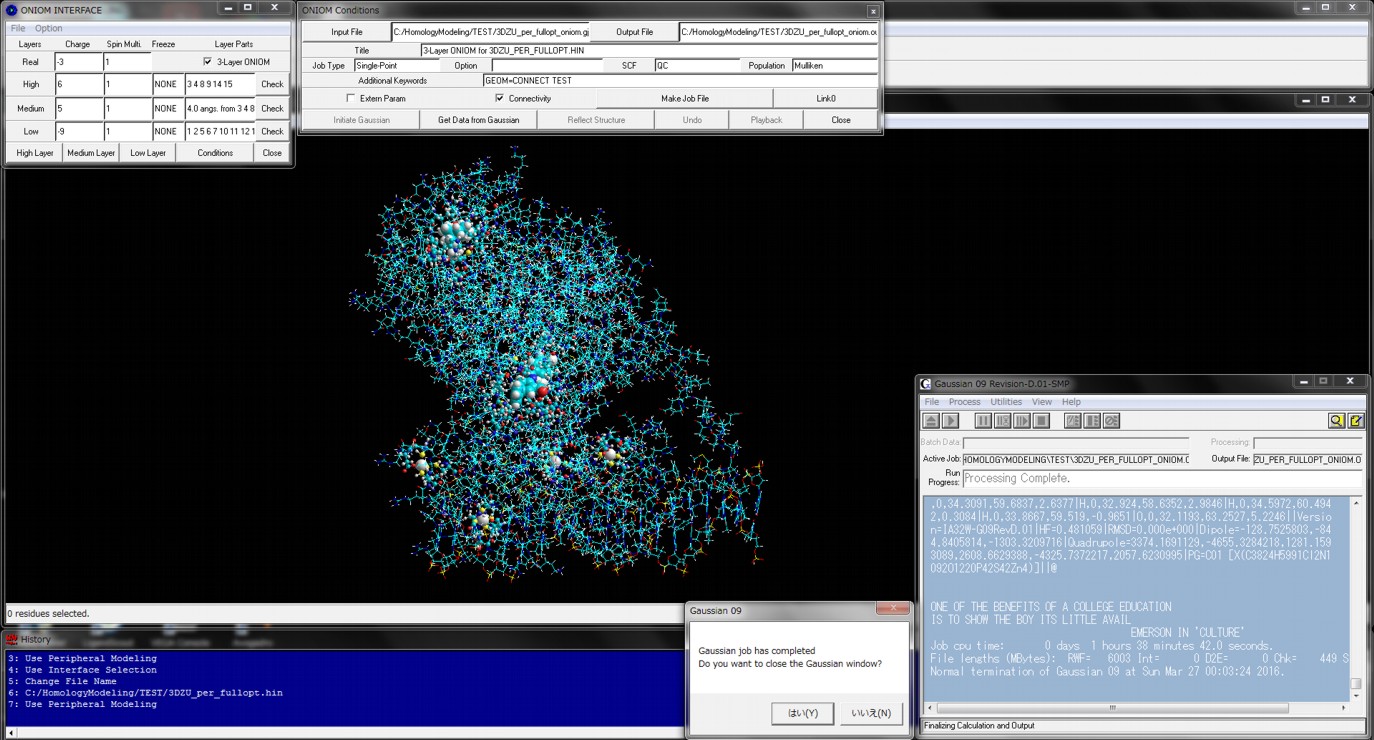
The following figure shows the result of ONIOM single-point calculations (RHF/6-31G:RAM1:AMBER) for the whole system which is constructed of a hetero dimer of two nuclear receptors (including ligand-binding domain and DNA-binding domain), hormone response elements of DNA, and cofactor fragments. In this calculation, two types of small compounds and four zinc atoms are set to the high layer (depicted in spheres), the protein residues, nucleotides, and water molecules around 4.0 angstrom from the high layer are set to the medium layer (depicted in balls and cylinders), and the other structures are set to the low layer (depicted in sticks). Since ONIOM Interface can automatically prepare all parameters, Gaussian program can be initiated simultaneously after starting the ONIOM Interface.
Added the Homology Search and Homology Modeling Reference manual.
Reference Manual Contents (Japanese Language)
Manual Contents (Japanese Language)
Homology Modeling Professional for HyperChem (PDF: 4 MB; Japanese)
SBDD Pamphlet (PDF: 2 MB; Japanese)
For early revisions:
What's New in Revision F2, 2012/01
What's New in Revision F1, 2010/02
What's New in Revision E1 and E2, 2008/10
What's New in Revision D1, 2008/05
What's New in Revision C2, 2007/11
What's New in Revision C1, 2007/02