

![]()
![]()
Last Modified
1 January 2024
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
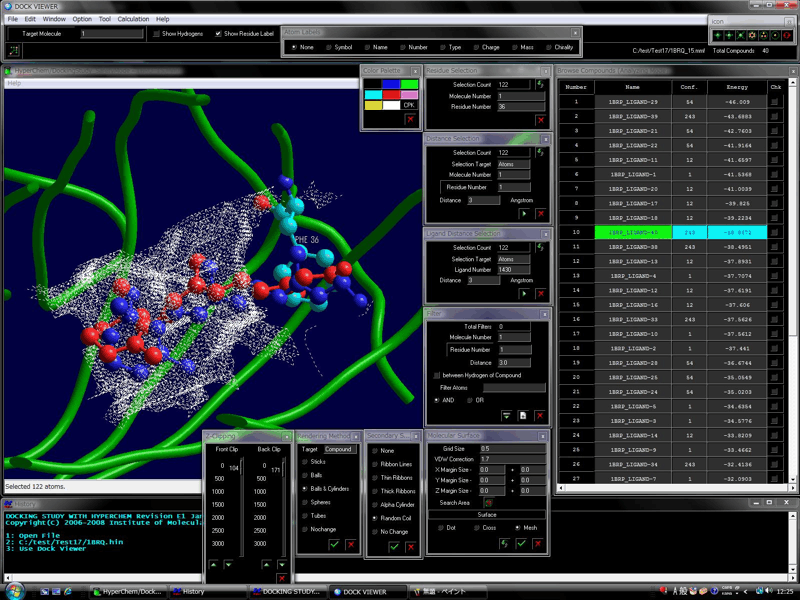
Docking Study with HyperChem
Essential, Premium Essential, Professional, Advanced, and Ultimate
What's New in Revision D1.
The full-automatic and comprehensive biological macromolecule (protein and nucleic acid molecule systems)- and ligand (small molecule and peptide)-flexible docking simulation program. It's easy to treat the comprehensive conformation search of compound, induced fit effects of the side chain and main chain of the biological macromolecule, and the flexibility of the other molecules such as water molecules, biological molecules, small molecules, and metal atoms, etc. The program (non-grid algorithm) can predict the precise interaction energy for the entire system rather than the approximated interaction energy obtained from the other docking simulation programs which adopt the grid algorithm for simplifying the energy calculations.
The following figure shows the results of the protein- and ligand-flexible docking simulations using the novel docking algorithm. In the apo conformation, the potential ligand-binding site is destroyed or occupied by its inner structure. The comprehensive and automatic docking simulations of the native ligand for the target adopting the apo conformation well reproduced the structure of the experimentally solved stable complex. In this figure, red shows the stable complex obtained from the docking simulations and blue shows the experimentally solved complex in the corresponding holo conformation.
The following shows the addition functions to the previous revision.
Supported the novel flexible docking algorithm which can be used for docking compound to a site in a large structural change exceeding the induced fit effect of the biological molecule such as the structural changes between the apo and holo conformations.
Transferred the common mathematical functions to the C++ dynamic link library (the program uses the HyperChem back-ended computational engines).
Supported the docking simulation with all pharmacophore points predicted on the basis of the structural manner.
Supported the simultaneous docking simulations at some docking sites.
The automatic biological macromolecule and peptide docking simulation is also available within an allowable simulation time.
Revised the User's manual (Japanese language).
Manual Contents (Japanese Language)
Homology Modeling for HyperChem
Homology Modeling Professional for HyperChem
What's New in Revision E1.
The protein homology modeling can be performed using only three module programs to optimize the structure of the main chain, side chain, and whole molecule system. Since these module programs can automatically operate, the homology modeling can be precisely performed under the almost full automatic conditions.
The program package can be used for performing the protein homology modeling in the presence or absence of the small molecules, water molecules, other biological molecules, metal atoms, and the small molecule covalently bound to the model under the vacuum or the various solvated conditions. The package also provides the several modeling, simulating, and analyzing functions.













The following shows the addition functions to the previous revision.
Sophisticated many functions carried on Revision C2.
Renewed the user interface of all module programs.
Window color can be selected from both the Windows default color and Dark color.
Confirmed stable performance for the molecular system exceeding 100,000 atoms (up to 65,000 atoms for single point and optimization calculation in HyperChem7.5.2 or later).
Transferred to the multiple window system from the modal window system.
Added the window management function.
Transferred the common mathematical functions to the C++ dynamic link library (the program uses the HyperChem back-ended computational engines).
Provided the homology search environment to the Control Center module program.
Added the Homology Search and Homology Modeling Reference manual.
Reference Manual Contents (Japanese Language)
Revised the User's manual (Japanese language).
Manual Contents (Japanese Language)
Homology Modeling Professional for HyperChem (PDF: 4 MB; Japanese)
SBDD Pamphlet (PDF: 2 MB; Japanese)
For early revisions:
What's New in Revision C2. 2007/11
What's New in Revision C1. 2007/02